Fig. 13.1
From barotrauma/volutrauma to biotrauma. Ventilator-induced lung injury progresses from the primary mechanical injury to secondary atelectrauma, encouraged by both barotrauma and volutrauma. Atelectrauma resulting from alveolar interdependence is showed in panel 3, where at the interface between collapsed/consolidated (A) and overdistended lung units, the tissue may be injured by excessive shear stress and stretching caused by the uneven expansion of surrounding zone (C). Panel 4 shows the stress concentration between an air-filled and edematous alveolus (a model of forces between air-filled and air-filled alveoli where all forces are in balance – A; a model of forces between an air-filled and edematous alveolus where a greater pressure drop across the alveolar interface causing and excessive strain). The progression of lung damage is seen through inflammatory mediators (panel 5), eventually resulting in distal tissue damage. Mechanical stretch impairs alveolar epithelial integrity, causing a loss of tight junction structure and cell attachment associated with a production of pro-inflammatory cytokines
Table 13.1
Summary of the most relevant studies regarding the ventilator-induced lung injury (VILI)
Concept | Author | Study type | Main thought |
|---|---|---|---|
Barotrauma/volutrauma, atelectrauma | Kumar et al. 1973 [35] | Prospective | IPPV + PEEP vs IPPV at ZEEP and development of barotrauma. Major determinant a preexisting COPD |
Uhlig et al. 2012 [9] | Overview | Discussion of the forces generated by MV and how they may injure the lungs mechanically and through inflammation | |
Lachmann et al. 1985 [33] | Experimental | A randomized study of five different ventilatory modes in a piglet model of severe respiratory distress | |
Tremblay et al. 1998 [27] | Overview | Description of the mechanisms of VILI, including the involvement of cellular and inflammatory mediator-induced injury | |
Ioannidis et al. 2015 [36] | Overview | Focus on barotrauma, from definition to treatment | |
Dreyfuss et al. 1988 [3] | Experimental | Comparison of the consequences of normal tidal volume ventilation in mechanically ventilated rats at a high airway pressure with those of high tidal volume ventilation, including the effects of PEEP on both edema and lung ultrastructure | |
Dos Santos et al. 1985 [39] | Overview | A perspective on ventilator-induced lung injury with a focus on mechanisms and clinical implications. | |
Valenza et al. 2003 [44] | Experimental | Investigation on the protective role of PEEP with respect to the time needed to reach similar levels of lung injury | |
Tremblay et al. 2005 [21] | Overview | From bench to bedside | |
Fanelli et al. 2009 [50] | Observational | Useful for assessing diaphragmatic motion under resistive loading | |
Stress, strain, stress raisers | Mead et al. 1970 [43] | Observational | Pioneeristic description of lung stretching forces distribution. |
Chiumello et al. 2008 [20] | Prospective | Plateau pressure and tidal volume are inadequate surrogates for stress and strain; quantification of the stress to strain relationship | |
Protti et al. 2011 [22] | Experimental | To identify a strain–stress threshold above which ventilator-induced lung damage can occur | |
Protti t al 2013 [23] | Experimental | To clarify whether different combinations of dynamic and static strains, resulting in the same large global strain, constantly produce lung edema | |
Protti et al. 2015 [24] | Experimental | To find the volumetric VILI threshold and see whether PEEP is protective per se or indirectly | |
Nieman et al. 2016 [28] | Overview | Using engineering concepts to analyze the impact of the mechanical breath on the lung is a novel new approach to investigate VILI mechanisms and to help design the optimally protective breath | |
Cressoni et al. 2014 [29] | Retrospective | To quantify lung inhomogeneities in patients with ARDS | |
Chiumello et al. 2016 [47] | Prospective | Airway pressures and tidal volume normalized to body weight as surrogates for lung stress and strain in mild pediatric ARDS | |
Biotrauma | Tremblay et al. 1997 [13] | Experimental | To study the effect of ventilation strategy on lung inflammatory mediators in the presence and absence of a preexisting inflammatory stimulus |
Muscedere et al. 1994 [42] | Experimental | To examine the hypothesis that ventilation at very low lung volumes can also worsen lung injury by repeated opening and closing of airway units as ventilation occurs from below to above the infection point as determined from the inspiratory pressure–volume curve | |
Ridge et al. 2005 [41] | Experimental | To demonstrate that shear stress causes disassembly of keratin in lung alveolar epithelial cells | |
Vlahakis et al. 2005 [40] | Overview | Experimental evidence for lung cells as injury targets and the relevance of these studies for human ventilator-associated lung injury |
13.6 Mechanical Power
Very recently, a new way of looking at the ventilator side of VILI, i.e., the mechanical power, has been introduced [18] . According to this approach, every ventilator component already known to be associated with the development of VILI (tidal volume, driving pressure, respiratory rate, inspiratory-to-expiratory ratio and flow), with the addition of the effect of PEEP, contributes, each one to his proper extent, to the total amount of energy delivered to the respiratory system (and hence to the lung). The mechanical power concept does not introduce any new component to the field of ventilator-related causes of VILI; instead, it proposes and validates a mathematical description of machine power responsive to the relative contributions of its bedside-adjustable components. Starting from the classical equation of motion, an equation was developed that enables to calculate the mechanical power by some easily obtainable ventilator variables [53]. In fact, the initial trigger of stress and strain is the force applied to the lung extracellular matrix times its displacement, which equals the product of pressure times delta-volume. The cyclic energy loads applied at a given frequency (power) then triggers the development of VILI, which may be seen in this context as a sort of “fatigue” of the extracellular matrix [24]. If the lung is subject to an “excess” of energy, the unrecovered energy may be expected to be sufficient to break the molecular bonds of the polymers of the extracellular matrix [54, 55], to detach endothelial [56] and epithelial [57] cells from the basement membrane, and to fracture the capillary walls [58]. Alteration of the extracellular matrix, combined with capillary micro-fractures, may then activate an inflammatory reaction [59] and micro-hemorrhage, leading to the extracellular edema typical of VILI.
Indeed, the tidal change in lung volume is associated with a cyclic energy load delivered by the ventilator to the respiratory system. The energy load to the respiratory system (Fig. 13.2) is composed of a static component, due to PEEP and PEEP volume (conceptually equivalent to potential energy), and a dynamic cyclic component, due to driving pressure and tidal volume above PEEP (conceptually equivalent to kinetic energy), plus the additional, resistive and inertial component generated by the pressure spent for gas movement, the surface tension forces, and tissue resistances to motion. Energy is equal to the pressure applied times the change in volume, summed along the inspiratory volume-pressure curve. In contrast, once PEEP is applied, no further cyclic energy load is imposed on the system, as the volume is constant. As a matter of fact, PEEP plays a complex role in the context of the energy provided by the ventilator, as it provides increased continuous tension to the extracellular matrix which then accumulates potential energy. Further energy is added when cyclic tidal ventilation is superimposed to reach a given end-inspiratory volume. Therefore, if the end-inspiratory volume is the same, with or without PEEP, the energy is lower in the presence of PEEP than without it. Computed in this way, the energy/power load provided a single explanation of the different phenomena related to development of VILI.
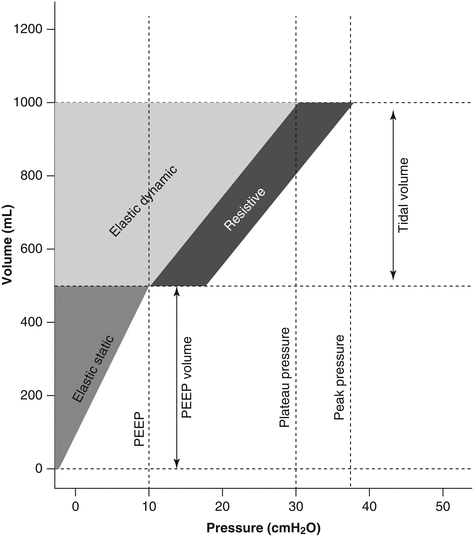
Fig. 13.2
Mechanical power and mechanical power equation. Mechanical energy provided during tidal ventilation and graphical representation of the power equation. The graphic is composed of a triangle on the lower left-hand side of the image, representing the elastic static component, i.e., the energy delivered just once when PEEP is applied, and of a larger trapezoid, representing the elastic dynamic component, whose area equals the elastic energy delivered at each tidal breath, to which a parallelogram is added on the right, representing the resistive component
In a recent paper, Gattinoni et al. [60] demonstrated how the “power equation,” as derived from the classical equation of motion with the addition of PEEP (while inertial forces were neglected), proved to yield comparable values of mechanical power when compared to data obtained experimentally through the pressure–volume curve analysis. The advantage of such a mathematical description of the mechanical power is that it enables the quantification of the relative contribution of its different components, thus allowing to anticipate the effects of their changes.
As far as mechanical ventilation is provided with PEEP, the static energy to reach the PEEP volume corresponds to the triangle equal to 1/2 × PEEP × PEEP volume. However, this energy is provided only once (as long as the same amount of PEEP is maintained), since during tidal ventilation the PEEP volume equals zero. However, in the presence of PEEP, more energy is required to inflate the lung. The energy needed for TV to reach P plat corresponds to the trapezoid equal to (peak pressure − PEEP) × TV/2 + (PEEP × TV).
From the classical equation of motion in which PEEP is also considered [61], at any given time, the pressure (P) in the whole respiratory system is equal to
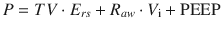 where E rs is the respiratory system elastance, R aw is the total respiratory system resistance, and V i is the inspiratory flow.
where E rs is the respiratory system elastance, R aw is the total respiratory system resistance, and V i is the inspiratory flow.
The energy provided by the ventilator per breath can be calculated by multiplying each pressure in the motion equation by the volume variation (i.e., TV); after substituting V i with TV/T insp (the inspiratory time) and then expressing T insp as a function of respiratory rate (RR) and inspiratory-to-expiratory (I:E) ratio and converting the value to J/min, the following equation may be derived:
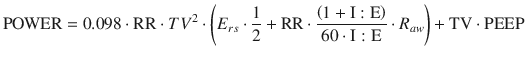
In the same paper [60], the authors demonstrated how, both in patients with healthy lungs and in patients with ARDS, the mechanical power measured directly through the pressure–volume curve analysis was strictly correlated with the one computed via the power equation.
Recently, in a secondary analysis of patients enrolled in two previously published randomized controlled trials [62], Guerin and colleagues [63] found how mechanical power (computed as DP × TV × RR, where DP is the airway driving pressure) was higher in ARDS non-survivors than in survivors. A dose–response effect was found, with higher values of power associated with increased mortality, and a threshold of 12 J/min was identified, which was associated with significant distinct probabilities of survival. Despite the equation proposed by the authors is simpler than that derived from the equation of motion, it represents only the product of respiratory rate times twice the dynamic energy component due to tidal volume, thus neglecting both the role of PEEP and that of the resistive load.
Since VILI originates from the interaction between the mechanical power transferred to the ventilable lung parenchyma and the anatomo-pathological characteristics of the latter, it is possible that different combinations of the components of mechanical power, resulting in a value greater than a given threshold, may produce similar damage. This was recently confirmed by animal experiments in which different combinations of tidal volume and respiratory rate were applied to detect the threshold for VILI [18]. The authors found how up to a mechanical power of approximately 12 J/min, the computed tomography scans showed mostly isolated densities, whereas when mechanical power was above the 12 J/min threshold, all piglets developed whole-lung edema.
Since airway pressure represents the pressure applied to the respiratory system as a whole, its interpretation is influenced by alterations in the mechanical properties of the chest wall [20, 64]; a more informative parameter could be the computation of the mechanical power selectively applied to the lung, either via the transpulmonary pressure–volume curve analysis or via a rearrangement of the equation of motion. The mechanical power delivered to the lung (POWERl) implies the use of the transpulmonary pressure (P l) instead of airway pressure at P plat and at PEEP. The relationship between P l and P aw (either P plat or PEEP) is expressed by P l = P aw × (E l/E rs), where E l is the elastance of the lung [65].
Therefore, substituting in the equation:
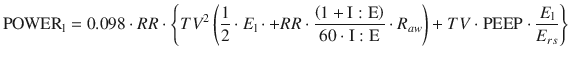
Eventually, it must be stressed that the mechanical power is just one part of the problem. The other part is represented by the lung’s conditions. The same mechanical power may have different effects depending on dimensions of the lung, the presence of inhomogeneity, the extent of the stress risers, and the vessels’ filling state, all factors which condition an uneven distribution of the delivered energy. Therefore, to be clinically meaningful, the mechanical power must be normalized, at least to the lung volume [66].
13.7 Extra-Parenchymal Factors
Nonmechanical background factors have repeatedly been proven important in the process of VILI generation, and, for any given ventilation pattern that applies potentially damaging stress, they may determine whether or not VILI is expressed [67]. These background factors, which may synergize with each other, include the preinjury and inflammatory state, the temperature at which ventilation occurs [68, 69], the amplitudes of vascular pressures and flows [56, 70], PaCO2/pH [71, 72], and FiO2 [73, 74].
In an experimental study aimed at investigating whether thermal stress could modulate the development of VILI, Suzuki et al. [68] randomized three groups of anesthetized rabbits to be ventilated for 2 h at core body temperatures of 33, 37, or 41° while ventilated with a noninjurious or a potentially injurious strategy. The hyperthermic group compared with the hypothermic animals developed a significantly higher degree of hypoxia and had increased lung edema and an altered pressure–volume relationship. To correct for potential effects arising from cardiac output fluctuations or from extrapulmonary organs, an isolated lung model was used for a confirmatory study, with similar findings. In a similarly designed study on rats, Akinci et al. [69] found how concomitant hyperthermia increased systemic inflammatory response, as assessed by higher levels of serum chemokines and cytokines, and a worse histology, during an injurious, high-pressure ventilation strategy.
Despite the great majority of studies of VILI which have specifically investigated the airspace mechanics, i.e., factors as tidal volume, plateau pressure, and PEEP, one must consider that the pulmonary alveolus is an interface between gas and blood. Indeed, because the intraluminal pressures applied to the airway epithelium also impact on the vascular endothelium, the potential for pressures and flows within blood vessels to influence the development and/or evolution of VILI has also been taken into account. The vascular structure of the pulmonary circulation is composed of both intra-alveolar and extra-alveolar vessels, whose behavior during lung inflation is fundamentally different. In fact, inflation compresses wall-embedded capillaries but dilates extra-alveolar microvessels. Under the high-permeability conditions of ARDS, even small increases in pulmonary microvascular pressure lead to increased edema formation. Moreover, unlike in health tissue in which the blood–gas barrier is intact, there is no clear pressure threshold for edema formation in the lung tissue of ARDS patients [75]. It is a well-known phenomenon that mechanical forces that tear the delicate alveolar–capillary membrane can originate on either side of the boundary, when the application of adverse ventilatory patterns to previously healthy lungs not only causes formation of proteinaceous edema, but it also stimulates neutrophil aggregation and hemorrhage [76]. The complementary issue, that is, the vascular contribution to VILI, was investigated in a series of studies conducted by the group of Marini using isolated, ventilated, and perfused lungs. When isolated rabbit lungs were exposed to perfusion levels that were equivalent to greater or less than the normal resting blood flow of in vivo animals, while ventilated with airway pressures that proved damaging in vivo, the authors demonstrated that perfusion amplitude contributed to the reduced lung compliance and promoted both lung edema and hemorrhage [77]. In a subsequent experiment [78], the authors found a significant relationship between the magnitude of pulmonary arterial pressure and the length of time over which it was sustained with the extent of VILI. Subsequent studies showed how lungs exposed to cyclic elevations in pulmonary artery pressure in the absence of ventilation formed less edema and exhibited less perivascular and alveolar hemorrhage than did ventilated lungs exposed to similar peak and mean pulmonary artery pressures and mean airway pressure [70]. Taken together, the studies demonstrated that when the mechanical stresses of the tidal cycle are high, an increase in precapillary vascular pressure or a reduction in postcapillary vascular pressure each could influence the severity of VILI inflicted by an unchanging adverse pattern of ventilation [56]. These observations imply that the gradient of transalveolar vascular pressure may be instrumental in inflicting damage when airway stresses are high. Figure 13.3 shows the possible mechanisms by which hemodynamic parameters may induce or exacerbate VILI.
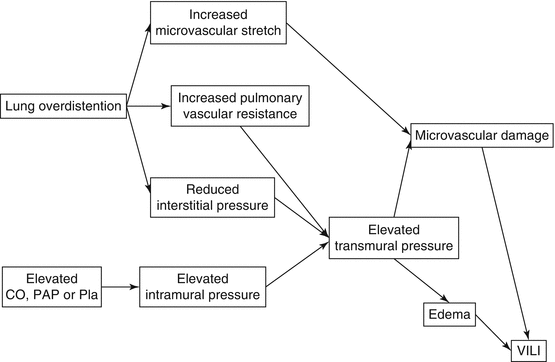
Fig. 13.3
Possible mechanisms by which pulmonary vascular hemodynamic parameters may incite or exacerbate ventilator-induced lung injury (VILI). Microvascular strain may be amplified at the junctions of open and closed lung units (CO cardiac output, Pla left atrial pressure, PAP pulmonary artery pressure)
In an attempt to reduce the total amount of stress delivered by the mechanical ventilator, a strategy of reduced minute ventilation has long ago been suggested [79], which was generally associated with increased carbon dioxide retention and hypercapnic acidosis. Indeed, experiments by Sinclair and coworkers [80] and by Broccard and colleagues [81] strongly indicate that the generation of hypercapnic acidosis may exert a protective effect on the severity of VILI. More recent preclinical data still confirm the protective effects of therapeutic hypercapnia initiated concurrently with injurious ventilation in an in vivo model of VILI [72]. The beneficial effects of such hypercapnic acidosis in reducing the extent of VILI are increasingly well understood and include attenuation of lung neutrophil recruitment, pulmonary and systemic cytokine and eicosanoid concentrations, cell apoptosis, and oxygen-derived and nitrogen-derived free radical injury [71].
The deleterious effects of hyperoxygenation are increasingly being recognized in several fields of intensive care medicine [82], to the extent that less aggressive approaches to oxygen supply have recently been suggested, even to not providing any supplemental oxygen, in several acute care settings, such as resuscitation of asphyxiated newborns, during acute myocardial infarction or after stroke or cardiac arrest [83]. Indeed, a recent trial showed how a conservative protocol for oxygen therapy vs conventional therapy resulted in lower mortality in a heterogeneous population of mechanically ventilated critically ill patients [84]. Experimental evidence points to the detrimental role of hyperoxia as a contributing factor to the development of VILI. In a laboratory investigation aimed at assessing whether hyperoxia could exacerbate lung injury caused by an injurious mechanical ventilation strategy, Sinclair and colleagues [73] found how hyperoxic animals, as compared to normoxic ones, had significantly reduced oxygenation and increased lung injury scores. Hyperoxia also significantly increased alveolar–capillary permeability and polymorphonuclear leukocytes and inflammatory mediator concentrations in bronchoalveolar lavage fluid. Similar results were found by Li et al. [74], who investigated the mechanisms regulating the interaction between injurious ventilation and hyperoxia. In their experiments, the authors found how the addition of hyperoxia to an unsafe ventilation strategy increased lung cytokine production, neutrophil infiltration by upregulation of the cytokine macrophage inflammatory protein-2, and apoptotic cell death through activation of the JNK and ERK1/2 pathways.
In conclusion, the vascular/metabolic/oxidative environment of the lung tissue at the onset of major mechanical stress has repeatedly shown to be influential in the VILI development process. Lowering ventilatory and cardiovascular demands – thereby reducing both minute ventilation and the vascular pressure gradient across the lung – are potentially important therapeutic targets when attempting to avoid VILI. It seems rational then to suggest a reduction in oxygen demand and ventilation requirement as a component of a comprehensive “lung-protective” strategy [67]. Indeed, reducing the ventilation requirement simultaneously allows reduction of driving pressures or ventilating frequencies. Because cardiac output also declines in response to lower oxygenation demands, the pulmonary microvascular blood flow gradient is lessened, thereby potentially reducing VILI risk.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


