Chapter 51 Ventilator-Induced Lung Injury
Mechanical ventilation is essential to life support of patients with respiratory failure. Several potential drawbacks to mechanical ventilation were identified early in its history.1 More recent experimental studies have showed that certain ventilation modalities may produce subtle tissue damage similar to that seen in early acute respiratory distress syndrome (ARDS) and termed ventilator-induced lung injury (VILI). This issue has recently received much attention in the clinical field.2,3 The purpose of this chapter is to describe pathophysiological events leading to VILI and to place these observations into a clinical perspective of ventilatory management of patients with ARDS.
Evidence for Ventilator-Induced Lung Injury
Ventilation of Intact Lungs
High Lung Volume Ventilator-Induced Lung Injury
Webb and Tierney4 found that pulmonary edema rapidly developed in rats subjected to 45 cm H2O peak airway pressure ventilation, whereas it did not develop in rats undergoing ventilation for a longer time with 14 cm H2O peak airway pressure. Edema severity and rate of development increased with peak airway pressure magnitude. It was later confirmed that such a ventilation modality produces endothelial and epithelial cell damage and lung capillary permeability changes that result in nonhydrostatic pulmonary edema.5 This edema formation is now generally believed to be the hallmark of VILI.
The respective roles of increased airway pressure and increased lung volume in this injury were clarified when mechanical ventilation at high and low tidal volume (VT) were compared at identical (45 cm H2O) peak airway pressures.6 The injury was found only in rats subjected to high VT and not in those undergoing ventilation at high airway pressure in which lung distention was limited by thoracoabdominal strapping (Figure 51-1).6 Furthermore, in animals undergoing ventilation at high VT by negative external distending pressure, pulmonary edema still developed, confirming that excessive airway pressure is not the causal factor of this type of injury.6 This VILI that depends mostly on end-inspiratory volume has been called volutrauma.7,8 The alveolar pressure corresponding to end-inspiratory volume is the “plateau” airway pressure (measured at no-flow), and its clinical importance has been emphasized in a Consensus Conference on mechanical ventilation.9 Several investigators reached the same conclusions in other species using different protocols.10–12
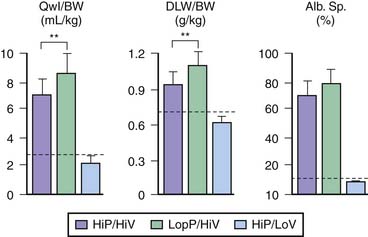
Figure 51–1 Comparisons of the effects of high-pressure–high-volume ventilation (HiP-HiV) with those of negative inspiratory airway pressure high tidal volume ventilation (iron lung ventilation, LoP-HiV) and of high-pressure–low-volume ventilation (thoracoabdominal strapping, HiP-LoV). Dotted lines represent the upper 95% confidence limit for control values. See Figure 51-3 for details on edema indices. Permeability edema occurred in both groups receiving high tidal volume ventilation. Animals undergoing ventilation with a high peak pressure and a normal tidal volume had no edema. BW, Body weight; DLW, dry lung weight; Qwl, extravascular lung water.
(From Dreyfuss D, Soler P, Basset G et al: High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure, Am Rev Respir Dis 137:1159-1164, 1988.)
Low Lung Volume Ventilator-Induced Lung Injury
Unlike volutrauma, ventilation at low lung volumes does not seem to injure healthy lungs. Intact animals tolerate mechanical ventilation with physiologic VT and low levels of positive end-expiratory pressure (PEEP) for prolonged periods of time without any apparent damage. Taskar and colleagues13 have shown that the repetitive collapse and reopening of terminal units during 1 hour of mechanical ventilation does not result in appreciable lung damage, although it does alter gas exchange and reduce compliance, as does spontaneous (low VT) ventilation under deep anesthesia.
Ventilation of Damaged Lungs
High-Volume Lung Injury
Several investigators have evaluated the effect of overdistension on damaged lungs. These studies consistently demonstrated the increased susceptibility of diseased lungs to the detrimental effects of some modalities of mechanical ventilation.
The first studies were performed on isolated lungs. Bowton and Kong14 showed that isolated perfused rabbit lungs injured by oleic acid gained significantly more weight when ventilated with 18 mL/kg VT than with 6 mL/kg VT. Hernandez and colleagues15 compared the effects of oleic acid alone, mechanical ventilation alone, and their combination on lung capillary filtration coefficient and wet-to-dry weight ratio (which reflects lung protein accumulation) in young rabbits. Filtration coefficient and wet-to-dry weight were not significantly affected by low doses of oleic acid or by mechanical ventilation at a peak inspiratory pressure of 25 cm H2O for 15 minutes. However, the filtration coefficient increased significantly when oleic acid injury was followed by mechanical ventilation at these pressures. Wet-to-dry weight ratio was also significantly higher than in lungs subjected to oleic acid injury or ventilation alone. The same team also showed that inactivating surfactant with dioctyl-succinate aggravates the filtration coefficient increase produced by ventilating isolated blood-perfused rabbit lungs at 30 to 45 cm H2O peak pressure.16 Whereas light microscope examination showed only minor abnormalities (minimal hemorrhage and vascular congestion) in the lungs of animals subjected to ventilation or surfactant inactivation alone, their combination caused severe damage (edema and flooding, hyaline membranes, and extensive alveolar hemorrhage).
These results suggested that ventilation at high volume and pressure might favor VILI in abnormal isolated lungs and that it might occur at lower airway pressure than in the normal lung. Whether this could also occur in lungs in situ was investigated by comparing the effects of different modalities of mechanical ventilation in rats with α-naphthylthiourea (ANTU)-injured lungs17 (Figure 51-2). ANTU infusion alone caused moderate permeability pulmonary edema. Mechanical ventilation alone resulted in a permeability edema of severity related to magnitude of VT. It was thus possible to calculate the theoretical amount of edema that would result from ventilating ANTU-diseased lungs with a given VT by summing up the separate effect of mechanical ventilation and ANTU. However, lungs of animals injured by ANTU had more edema than predicted when they underwent ventilation with a high VT (45 mL/kg body weight), indicating that the two insults acted in synergy. Even slight lung alterations, such as those produced by spontaneous ventilation during prolonged anesthesia (which inactivates surfactant and promotes focal atelectasis18,19), are sufficient to exacerbate the harmful effects of high-volume ventilation.17 The extent to which lung mechanical properties are altered prior to ventilation is a key factor in this synergy. The amount of pulmonary edema produced by high-volume mechanical ventilation in animals given ANTU, or after prolonged anesthesia, was inversely proportional to respiratory system compliance measured at the very beginning of high-volume mechanical ventilation.17 Thus the more severe the lung abnormalities were before ventilation, the more severe was the VILI.
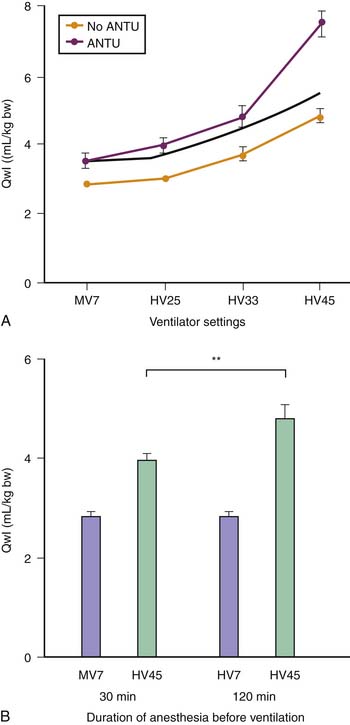
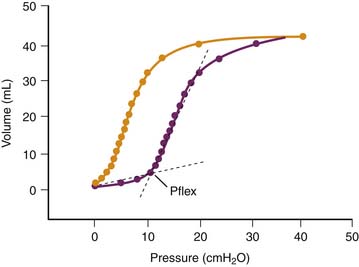
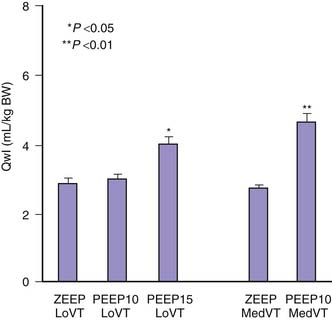
The reason for this synergy requires clarification. The presence of zones of alveolar edema in animals given this harmful ventilation was the most evident difference from those that underwent ventilation with lower, less harmful VT.17 Because alveolar flooding reduced the number of alveoli available for ventilation, they were more prone to overinflation, more vulnerable, and at greater risk of alveolar flooding. This in turn would further reduce aerated lung volume and result in positive feedback. The same reasoning applies to prolonged anesthesia, during which aerated lung volume was probably gradually reduced by atelectasis.17 Both flooding and atelectasis decrease lung compliance by a “baby lung” effect, a reduction in the volume of lung available for ventilation. It is not surprising that the lower the compliance before ventilation, the more severe were the lung alterations induced by high-volume ventilation.17 Thus uneven distribution of ventilation during acute lung injury20 favors regional overinflation and injury. To substantiate this phenomenon, rats underwent ventilation with VT of up to 33 mL/kg after alveolar flooding by instillation of saline solution into the trachea. Flooding with saline solution did not significantly affect microvascular permeability when VT was low. As expected, capillary permeability alterations were more important in animals that experienced alveolar flooding than in intact animals that underwent ventilation at high VT. Correlations also were found between end-inspiratory (plateau) airway pressure, the pressure at the “lower inflection point” on the pressure-volume (PV) curve, and the capillary permeability changes found in animals that experienced alveolar flooding and underwent ventilation with at high VT (Figure 51-3).21 Thus the changes in capillary permeability caused by lung overinflation are more severe in poorly recruitable (and less compliant) lungs.
Low-Volume Lung Injury
An increase in trapped-gas volume during pulmonary edema and acute lung injury probably occurs because of airway closure and is worsened by impaired surfactant function.22 Under such conditions, the slope of the inspiratory limb of the respiratory system PV curve often displays a sharp increase at low lung volume. This change reflects the sudden and massive opening of units previously excluded from ventilation and has been termed the “lower inflection point.” This phenomenon often has a dramatic effect on arterial oxygenation, because setting PEEP above the pressure at this inflection point often decreases shunt and increases PaO2.23–26
The possibility that pulmonary dysfunction may be aggravated if this inflection point lies within the VT has been a recent focus of attention. Experimental evidence for this possibility initially was provided by studies comparing conventional mechanical ventilation with high-frequency oscillatory ventilation in premature or surfactant-depleted lungs. Studies performed on such lungs ventilated at various levels of PEEP suggest that repeated closure and reopening of terminal units can cause lung injury.27–29 Argiras and colleagues27 and Sandhar and associates28 studied this issue in rabbits with surfactant-depleted lungs. During volume-controlled ventilation, peak inspiratory pressure was initially 15 mm Hg but increased to 25 mm Hg 5 hours later because lung compliance fell. PEEP was then adjusted to be either above (8 to 12 mm Hg) or below (1 to 2 mm Hg) the lower inflection point of the inspiratory limb of the PV curve. Mortality rates in the two groups were identical, but arterial PaO2 was better preserved and less hyaline membrane formation occurred in the high-PEEP group.27,28 This lessening of pathologic alterations was observed even when inspiratory/expiratory time ratios were adjusted so that mean airway pressures were the same in the low-PEEP and high-PEEP groups.28 Muscedere and colleagues30 reported similar results in isolated, unperfused rabbit lungs lavaged with saline solution and ventilated with a low (5 to 6 mL/kg) VT and with a PEEP set below or above the inflection point. However, Sohma and colleagues29 did not find this injury in vivo in rabbits whose lungs had been injured with hydrochloric acid.29
It often is argued that the lower inflection point on the PV curve reflects the recruitment of collapsed zones that are found predominantly in dependent areas31,32 and that this recruitment persists during further lung expansion. Recently Martynowicz and coworkers33 have questioned the existence of the repetitive collapse-reexpansion phenomenon during tidal ventilation and have reevaluated the significance of the lower inflection point on the PV curve. They studied the regional expansion of oleic acid–injured lungs using the parenchymal marker technique in dogs. They found that the gravitational distribution of volume at functional residual capacity was not affected and not associated with a decreased parenchymal volume of the dependent regions. In addition, they found that the between-region asynchrony of tidal expansion was not influenced by oleic acid injury. Their findings therefore did not support the hypothesis that a more important gravitational gradient during VILI produces atelectasis by compression of the dependent lung, cyclic recruitment and collapse, and ultimately shear stress injury.33 They propose that the displacement of air-liquid interfaces along the tracheobronchial tree causes the lower inflection point on the PV curve and conclude that this knee on the curve reflects the mechanics of partially fluid-filled alveoli with constant surface tension and not the abrupt opening of airways or atelectatic parenchyma.34 It therefore remains unsettled whether injury caused by the repetitive reopening of collapsed terminal units and the protective effect of PEEP is restricted to the peculiar situation of surfactant depletion by bronchoalveolar lavage. In the clinical arena, the recent negative results of the ALVEOLI trial35 and the two other more recent randomized controlled trials36,37 cast doubt on the clinical existence of repetitive opening and closing lung injury.38
Roles of Tidal Volume, Positive End-Expiratory Pressure, and Overall Lung Distention
Effects of Positive End-Expiratory Pressure When Tidal Volume Is Kept Constant
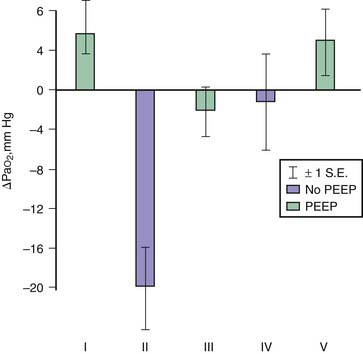
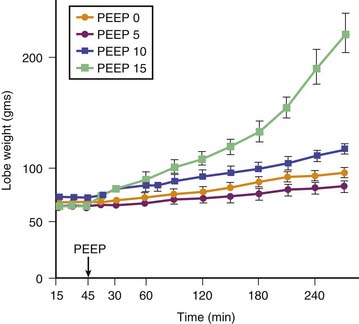
Application of PEEP may result in lung overinflation if it is followed by a significant change in FRC because of the increase in end-inspiratory volume. Overinflation will affect preferentially the more distensible areas that receive the bulk of ventilation, which may explain the lack of reduction or even the worsening of edema reported following PEEP application in most experiments.39 PEEP does not affect the severity of hydrostatic40 or permeability40,41 edema in intact animals, although it improves oxygenation40 because of the recruitment of flooded alveoli (Figure 51-4). In isolated ventilated-perfused lungs, PEEP rather aggravates edema fluid accumulation42 (Figure 51-5). Thus when VT is left unchanged, increasing FRC with PEEP affects edema differently in isolated lungs or in intact animals. In the latter, the lack of effect of PEEP depends on the balance between PEEP-induced increase in end-inspiratory lung volume, which decreases interstitial pressure and increases filtration pressure in extra-alveolar vessels, and the hemodynamic depression due to elevated intrathoracic pressure that, in the opposite, decreases filtration pressure. In contrast, preservation of perfusion rate in isolated-perfused lungs favors edema formation.42
Effects of Positive End-Expiratory Pressure when Tidal Volume Is Reduced
Edema is less severe during high-volume ventilation (even though FRC is increased by PEEP) when end-inspiratory lung volume is kept constant by decreasing VT (Figure 51-6). Webb and Tierney4 showed that less edema developed during ventilation with 45 cm H2O peak airway pressure when 10 cm H2O PEEP was applied. The authors attributed this beneficial effect of PEEP to the preservation of surfactant activity. It was later shown that, although PEEP decreased the amount of edema, it did not prevent alteration of capillary permeability.6 However, animals undergoing ventilation with PEEP had no alveolar damage in contrast with those that underwent ventilation without PEEP. The only cellular alterations found in animals that underwent ventilation with PEEP consisted of capillary endothelial blebs.6 No satisfactory explanation has been found for this preservation of the epithelial layer. It may be that PEEP prevented fluid movement in terminal units, thereby decreasing shear stress at this level. Similar observations have been made by other investigators in intact animals43,44 and in isolated perfused canine lobes.45 The hemodynamic alterations induced by PEEP probably play an important role in lessening the severity of edema. Application of PEEP produces an increase in mean intrathoracic pressure that adversely affects cardiac output.46,47 For example, in rats subjected to high peak airway pressure and 10 cm H2O PEEP, edema was more severe when the hemodynamic alterations induced by PEEP were corrected with dopamine infusion.48 The amount of extravascular lung water was correlated with systemic blood pressure, suggesting that restoration of cardiac output increased filtration pressure and was responsible for aggravation of edema. The reduction of edema and of the severity of cellular damage by PEEP during ventilation-induced pulmonary edema may be linked to reduced tissue stress by decreasing volume-pressure excursion, movement of foam in distal airways, preservation of surfactant activity, and a decrease in capillary filtration.
< div class='tao-gold-member'>





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


