Ventilator-Induced Lung Injury: Introduction
The deleterious effects of mechanical ventilation on the lungs have long been referred to as barotrauma. For many years, clinicians defined barotrauma as the occurrence of air leaks resulting in the accumulation of extraalveolar air responsible for a number of manifestations, of which the most threatening is tension pneumothorax. In addition to these “macroscopic” events whose adverse consequences are usually immediately obvious, mechanical ventilation may produce more subtle physiologic and morphologic alterations, especially when it results in high airway pressures. Our knowledge of such alterations has stemmed mainly from experimental studies and has expanded considerably over recent years. Indeed, alterations in alveolar–capillary barrier integrity and release of both inflammatory and antiinflammatory mediators have been reported in animals ventilated with modalities resulting in high lung stretching. Tissue damage also may occur during mechanical ventilation when distal airways close and open repeatedly because of the movement of foam in the airway lumen or rupture of liquid menisci. The clinical relevance of these experimental findings received resounding confirmation with the results of the ARDS Network study, which showed a 22% reduction in mortality in patients with the acute respiratory distress syndrome through a simple reduction in tidal volume.1
The first comprehensive work demonstrating that mechanical ventilation may be unsafe in intact animals was performed by Webb and Tierney.2 Rats ventilated with a peak inspiratory pressure (PIP) of 30 or 45 cm H2O displayed pulmonary edema within 20 minutes to 1 hour, depending on the pressure level. Microscopic examination of the lungs disclosed moderate interstitial edema in animals ventilated with the lower peak pressure, contrasting with profuse edema and alveolar flooding in animals ventilated with the highest PIP. Other studies subsequently documented the occurrence of pulmonary edema and lung ultrastructural abnormalities3,4 after even very short periods of intermittent positive-inspiratory pressure with high PIP. Kolobow et al5 reported progressive lung injury in sheep ventilated with 50 cm H2O PIP over 48 hours, manifested as decreased pulmonary compliance and deterioration in blood oxygenation. Some of the animals died before the 48-hour end point. At autopsy, lungs exhibited congestion and severe atelectasis.
The mechanisms underlying this ventilator-induced lung injury (VILI) have been for the most part elucidated. Two main factors explain its development: the magnitude of lung overdistension and its duration. Ventilation with very high peak transalveolar pressure results in acute, rapidly fatal, permeability pulmonary edema, whereas more protracted ventilation involving alveolar distension of lesser magnitude produces a lung injury in which inflammatory phenomena may play a role. This chapter presents the current knowledge on VILI. A Medline research employing “ventilator-induced lung injury” as keywords and limited to English articles yielded approximately 1900 results up to January 2005 (when the chapter in the second edition of this book was written). A Medline search employing the same keywords yielded 3860 results when updated to December 2010, reflecting the huge scientific productivity in this field over the past 6 years. To provide the most recent findings to readers, we have condensed material that was presented in greater detail in the second edition of this book.
Mechanisms of Ventilator-Induced Lung Injury
Ventilation modalities with very high distending pressures (typically >30 cm H2O, depending on the species) result in pulmonary edema. Both increased filtration and alteration in capillary permeability participate in edema formation. The reasons for these abnormalities have been partly elucidated.
The first hypothesis put forward to account for ventilator-induced edema involved hydrostatic alterations.2,6 Parker et al6 calculated that mean lung microvascular pressure increased by 12.5 cm H2O during ventilation of open-chest dogs at 64 cm H2O PIP. Increased filtration may result from an increase of pulmonary capillary pressure, a decrease in lung interstitial pressure, or both. Surfactant inactivation and increased lung volume participate in the decrease in lung interstitial pressure.
The increase in alveolar surface tension resulting from surfactant inactivation would be expected to further decrease the negative pressures surrounding alveolar vessels, thereby increasing vascular transmural pressure and enhancing fluid filtration. Faridy et al7 showed that ventilating excised dog lungs altered pulmonary pressure–volume curves and increased surface tension of lung extracts commensurately with the magnitude of tidal volume and duration of ventilation. Comparable findings were reported in excised rat lungs.8,9 These anomalies were ascribed to depletion or inactivation of surfactant. Of note, surfactant alterations failed to occur when positive end-expiratory pressure (PEEP) was used.7,8,10,11
Pattle12 and Clements13 suggested that an increase in alveolar surface tension might increase filtration. Albert et al14 found that the isogravimetric pressure (the vascular pressure at which net fluid flux from pulmonary vessels is zero) decreased when alveolar surface tension was increased by cooling and ventilating lungs at a low resting volume in open chest dogs. The authors interpreted this fall in isogravimetric pressure as indicative of a fall in perimicrovascular pressure.
Aerosolized 99mTc-DTPA (technetium-99m diethylenetriamine pentaacetic acid) clearance was found to increase following detergent aerosolization in rabbits15 and dogs.16 This finding was ascribed to regional overexpansion secondary to uneven lung inflation during mechanical ventilation of alveoli with altered surface tension rather than to elimination of peculiar barrier properties of surfactant.16
Increased filtration during mechanical ventilation probably also occurs at the extraalveolar level. During lung inflation, because of “pulmonary interdependence,” the pressure in the perivascular space surrounding extraalveolar vessels decreases, which, in turn, increases transmural pressure. This effect of lung volume on pulmonary vessels is well documented.17 Inflating the lungs dilates the extraalveolar vessels.18 During inflation from a low transpulmonary pressure, the increase in vessel diameter is such that an effective outward-acting pressure in excess of pleural pressure (1 to 2 cm H2O for each centimeter of water increase in transpulmonary pressure) expands the vessels.19 The potential importance of fluid leakage through extraalveolar vessels has been established in both excised lungs20 and in situ lungs of open-chest animals.21 Moreover, inflation of in situ lobes under zone 1 conditions (i.e., where alveolar pressure exceeds pulmonary arterial and venous pressures) was found to precipitate hydrostatic pulmonary edema in dogs.22 Because there is no flow in capillaries under zone 1 conditions, it is likely that the edema fluid leaked from distended extraalveolar vessels. The rate of edema formation was significantly correlated with the level of alveolar pressure and, therefore, the magnitude of distension (Fig. 42-1).
Figure 42-1
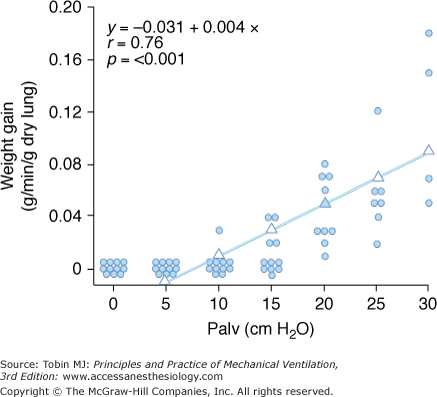
Relationship between alveolar pressure (Palv) and rate of hydrostatic edema formation in in situ lobes of open-chest dogs. Pulmonary arterial and venous pressures were kept at 1 cm H2O. When lung volume was low (Palv = 10 cm H2O), no edema occurred, whereas greater distensions produced a linear increase in lung weight gain, indicating edema formation. (Used, with permission, from Albert et al.22)
The effects of decreased perimicrovascular pressure and surfactant alterations probably combine with increased intravascular pressure to augment transmural microvascular pressure during overinflation. Although the magnitude of these changes is difficult to evaluate with precision, the increase in vascular pressure seems relatively modest and is unlikely to explain per se the rapid development of profuse edema (especially in small animals) observed after high PIP ventilation.
Many experimental studies in isolated lungs, as well as in open-chest or intact animals, have demonstrated permeability alterations (in response to high airway pressures) in the epithelium and, more unexpectedly, the endothelium.
The increase in alveolar epithelial permeability to small hydrophilic solutes observed when lung volume increases is a physiologic phenomenon. Elevation of functional residual capacity (FRC), in sheep, obtained by increasing the level of PEEP during mechanical ventilation23 or spontaneous ventilation,24 was associated with an increase in aerosolized diethylenetriamine pentaacetic acid (DTPA) clearance. Clearance augmentation was larger than expected from the changes in alveolar exchange surface area.
Effects of overinflation on epithelial permeability were studied extensively by Egan during static inflation of fluid filled in situ lobes.25,26 The equivalent-pore approach was used to describe the permeability of the epithelium to hydrophilic solutes of various sizes. Equivalent-pore radii increased from about 1 nm at 20 cm H2O inflating pressure to 5 nm at 40 cm H2O alveolar pressure. In some instances, free diffusion of albumin across the epithelium was observed, indicating the presence of large leaks (Fig. 42-2). Permeability alterations persisted or even increased after cessation of inflation, suggesting irreversible epithelial injury. High airway pressures applied to in situ lobes, however, resulted in supraphysiologic overinflation because of the compression of adjacent parenchyma. Egan27 performed experiments under less extreme conditions in rabbits, in which both in situ lobes and entire lungs were distended with 40 cm H2O airway pressure. Static segmental inflation resulted in a sixfold to 12-fold increase in lung volume from FRC and in an epithelium permeable to solutes such as albumin, cytochrome c, and cyanocobalamin. In contrast, whole-lung inflation resulted in only a threefold to fourfold increase in lung volume and a lesser rate of escape of the smaller solutes from the alveolar spaces. The increase in albumin permeability resulting from whole-lung overinflation was negligible. Hence, only major increases in lung volume produce significant changes in permeability to large molecules.
Figure 42-2
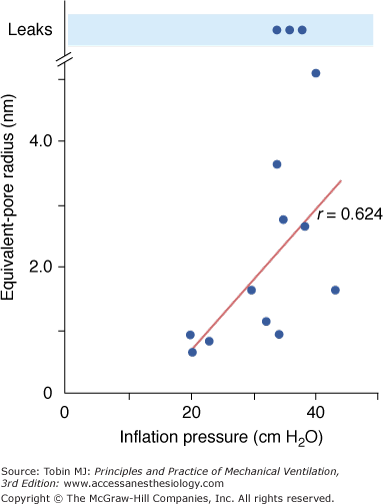
Effect of inflation pressure on the epithelial permeability of fluid-filled in situ lobes of sheep lung. Permeability is characterized by an equivalent-pore radius. A linear relationship was observed between inflation pressure and pore radius. At the highest levels of inflation, free diffusion of albumin was sometimes observed, indicating the presence of large leaks. (Used, with permission, from Egan et al.25)
Although less-well-documented on a quantitative basis, epithelial alveolar permeability alterations are probably present in varying degrees during ventilator-induced pulmonary edema. During positive pressure-ventilation with 41 cm H2O PIP, increases in alveolar permeability to small (DTPA), but not large (albumin), solutes were observed after 8 hours by Ramanathan et al in lambs.28 After 2 minutes of high-pressure ventilation, epithelial lining fluid (ELF) volume calculated from bronchoalveolar lavage increased by 180%4 (Fig. 42-3). ELF protein concentration decreased (Fig. 42-4A) even though protein content increased by 76% (Fig. 42-4B), suggesting that most of the excess fluid entered the alveolar-airway lumen through increased convection. In contrast to the severely altered sieving properties of microvessels after only 2 minutes of overinflation, epithelial permeability appears to be better preserved. This speculation is consistent with the scarcity of epithelial cell alterations on electron microscopic examination, contrasting with the widespread endothelial abnormalities.4 The presence of blood cells in the alveoli, however, suggested a few large endothelial and epithelial tears. During longer (2 hours) ventilation periods with a lower tidal volume (VT) (19 mL/kg in rats, resulting in a PIP of 19 cm H2O), the presence of secretory Clara-cell protein (a protein found in airway lumen) in the vasculature contrasting with a decrease in its concentration in bronchoalveolar lavage fluid further suggests an increase in alveolar barrier permeability.29 Secretory Clara-cell protein was also found in the plasma of mice ventilated with 35 cm H2O PIP for 2 hours and even earlier (30 minutes) when PIP was 55 cm H2O.30 Resorption of alveolar liquid by distal airway epithelium is decreased during VILI because of depressed sodium transport mechanisms.31 It can be restored by β-adrenergic stimulation that recruits ion-transporting proteins to the plasma membrane of alveolar epithelial cells or by β1-sodium-potassium adenosine triphosphatase (ATPase) subunit gene transfer.32,33
Figure 42-3
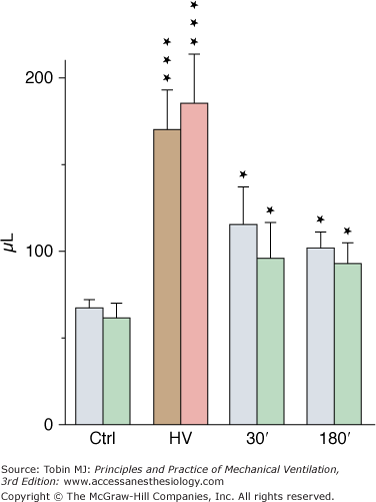
Estimates of epithelial lining fluid (ELF) volume by bronchoalveolar lavage after ventilation with 45 cm H2O peak airway pressure for 2 minutes (HV) and recovery. Two successive lavages were performed (grey and brown bars, first lavage; green and pink bars, second lavage). ELF volume increased significantly after HV and then decreased during recovery but remained higher than in control subjects (Ctrl). * = p < 0.05; *** = p < 0.001, as compared to controls. (Used, with permission, from Dreyfuss et al.4)
Figure 42-4
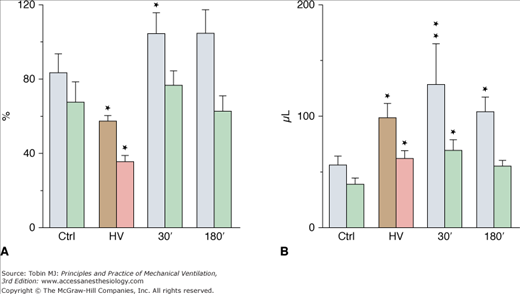
A. Protein concentration and (B) protein content in epithelial lining fluid (see Fig. 42-3 for details on the lavages). Protein content (expressed as the equivalent plasma volume) increased after high-pressure ventilation (HV) and then remained unchanged during recovery. Protein concentration (as a percentage of plasma protein concentration) decreased after HV and returned to normal during recovery (see text for details). * = p < 0.05; ** = p < 0.01, as compared to controls. (Used, with permission, from Dreyfuss et al.4)
In a study in isolated blood-perfused lobes from dogs, Parker et al34 demonstrated that ventilation for 20 minutes with graded increases in PIP did not affect microvascular permeability up to 30 cm H2O. Higher PIP (45 to 65 cm H2O) was associated with increases in the capillary filtration coefficient (Fig. 42-5) and decreases in isogravimetric capillary pressure, suggesting the existence of an airway pressure threshold. Similarly, the estimated protein reflection coefficient was decreased only at the highest airway pressures. Of note, at an airway pressure above the threshold, the increase in capillary filtration coefficient occurred immediately and continued in some lobes after cessation of distension.
Figure 42-5
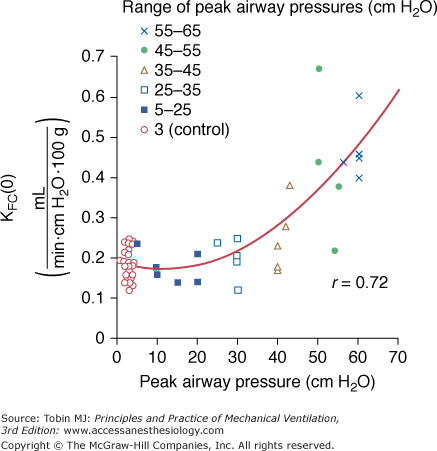
Effect of peak airway pressure on the capillary filtration coefficient (KFC) of isolated, blood-perfused lobes of dog lungs receiving 20 minutes of intermittent positive-pressure ventilation. Moderate (up to 30 cm H2O) increases in peak pressure did not affect KFC, whereas higher levels of peak pressure resulted in a very steep increase in KFC. (Used, with permission, from Parker et al.34)
To evaluate permeability alterations in intact animals, extravascular lung water and bloodless dry-lung weight and the distribution space in lungs of 125I-labeled albumin injected in the systemic circulation were measured in intact rats subjected to 45 cm H2O PIP ventilation.3 Pulmonary edema developed very rapidly and was easily demonstrable after only 5 to 10 minutes of high-pressure ventilation (Fig. 42-6). Light microscopic examination revealed that, at this stage, edema fluid remained confined to interstitial spaces, where it accumulated in the large peribronchovascular cuffs. No alveolar flooding was apparent. The presence of permeability alterations was indicated by a significant increase in dry-lung weight and of albumin distribution space (Fig. 42-6). After 10 minutes of high PIP ventilation, pulmonary edema was more abundant but still involved only the interstitial spaces. After 20 minutes of ventilation, findings were strikingly different, with tracheal flooding in all animals. Widespread alveolar flooding was obvious upon light microscopic examination. The severity of the permeability alterations was such that the ratio of 125I-albumin activity in tracheal fluid versus plasma was close to unity, indicating the loss of permselectivity of the capillary barrier. The severity of the permeability defect was indicated by the relationship between dry-lung weight and extravascular lung water (Fig. 42-7), which indicated that the concentration of protein in extravasated fluid was high, suggesting the presence of numerous large capillary leaks. Similar findings relating the duration of high-PIP ventilation and alterations in capillary permeability have been made in mice.30 This increase in endothelium permeability occurs both at the alveolar and extraalveolar level.35
Figure 42-6
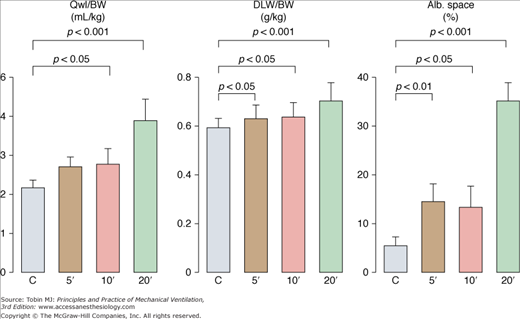
Effect of 45-cm H2O peak airway pressure ventilation in intact rats. Pulmonary edema was assessed by the determination of extravascular lung water content (Qwl/BW) and permeability alterations by the determination of bloodless dry lung weight (DLW/BW) and of the distribution space in the lungs of 125I-labeled albumin (Alb. Space). Permeability pulmonary edema developed rapidly (5 minutes). After 20 minutes of mechanical ventilation, there was a dramatic increase in all indexes (p < 0.01 versus other groups). (Adapted, with permission, from Dreyfuss et al.3)
Figure 42-7
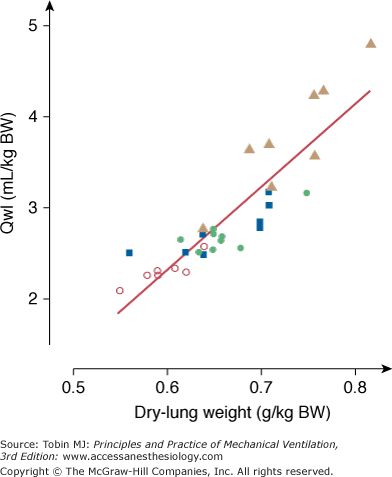
Relationship between extravascular lung water content (Qwl/BW) and dry lung weight during mechanical ventilation with peak airway pressure 45 cm H2O. The concentration of proteins in the edema fluid estimated from this relationship was consistent with the outpouring of protein-rich edema fluid reflecting severe permeability alterations. Open circles, control conditions; closed circles, 5 minutes of ventilation; closed squares, 10 minutes of ventilation; closed triangles, 20 minutes of ventilation. (Used, with permission, from Dreyfuss et al.3)
The time course of edema development depends on the size of the species: in rats, high PIP ventilation for as little as 2 minutes is sufficient to produce permeability edema,4 although alterations are minor and result in fairly mild edema. The increase in the ratio of extravascular lung water to blood-free dry-lung weight was increased by only 17% after 2 minutes4 versus 90% when the duration of the challenge was increased up to 20 minutes.36 In larger animals, considerably longer durations of high PIP ventilation are required to produce significant alterations. For instance, in lambs mechanically ventilated at 58 cm H2O PIP for 6 hours, Carlton et al37 found an increase in the ratio of extravascular lung water to dry-lung weight of only 19%. Results of histologic lung studies were either normal or showed only mild perivascular edema with no alveolar edema. In a study by Borelli et al,38 sheep mechanically ventilated for 18 hours with a PIP of 50 cm H2O developed both interstitial and alveolar edema.
Wet-lung weight normalized for body weight increased by 89% compared to normal lungs.39 After 27 hours of ventilation, pathologic abnormalities were much more marked, and lung weight was increased by 136% compared to normal lungs. Clearly, the ventilation time needed to produce severe edema is less than 1 hour in small species but more than 24 hours in large animals. Blood-gas barrier thickness is an important component of capillary resistance to mechanical stretch, and is more important in larger animals.40 Thus, one reason why permeability alterations were not consistently found is that they may become detectable only after longer ventilation times, in contrast to the immediate occurrence of microvascular pressure anomalies.
In summary, increased microvascular filtration pressure and altered microvascular permeability probably compound their effects to produce high PIP pulmonary edema. Although the hydrostatic component seems to be moderate on a quantitative basis, at least in closed-chest animals, it may nevertheless have a substantial impact. Indeed, in the face of a microvascular barrier with altered sieving properties, any increase in driving pressure will have a dramatic effect on edema formation.41–43
Interest has increasingly focused on the cellular response to mechanical strain and this subject has been comprehensively reviewed.44 Tschumperlin and Margulies45 observed increased cell death when alveolar epithelial cells were submitted in vitro to deformation. Cyclic deformation led to significantly greater cell death than did static deformation. The percentage of dead cells after 1 day ranged from 0.5% to 72% depending on the changes in surface area (up to 50%). In cyclically deformed cells, injury occurred rapidly, with most of cell death occurring during the first 5 minutes of deformation.46 These authors showed that basement membrane surface area increased approximately 40% when lung volume was varied from FRC to total lung capacity.47 These increases suggest that epithelial cells undergo significant stretch during large inflations. Vlahakis et al48,49 labeled membrane lipids to study deformation-induced lipid trafficking and studied in a direct manner (laser confocal microscopy) the response of epithelial cells of the alveolar basement membrane response to forces. A 25% stretch deformation resulted in lipid transport to the plasma membrane that allowed maintenance of its integrity and an increase in epithelial cell surface area. Such lipid trafficking occurred in all cells, whereas plasma breaks were seen in only a small percentage of cells. The authors concluded that deformation-induced lipid trafficking serves, in part, to repair plasma breaks so as to maintain plasma membrane integrity and cell viability, and that this could be viewed as a cytoprotective mechanism against the plasma membrane stress failure seen during VILI.36,50
Overinflating the lungs results in the increase in epithelial25 and endothelial3,34 permeability, both occurring after inflating the lung above the same end-inspiratory pressure threshold.51 Using an isolated perfused rat model, Parker et al showed that gadolinium (which blocks stretch-activated nonselective cation channels) annulled the increase in microvascular permeability induced by high PIP.52 This suggests that entry through stretch-activated channels and an increase in intracellular calcium ion (Ca2+) concentration might initiate the increase in permeability. This increase results in the activation of tyrosine kinases,53 activation of the Ca2+–calmodulin pathway and phosphorylation of myosin light chain kinase.54 Taken together, these results suggest that the increase in microvascular permeability below the cell rupture point, which occurs under extreme stretch conditions (“stress failure”50,55), is not simply a passive physical phenomenon, but the result of biochemical reactions. In vitro studies show that cell plasticity and deformation-induced lipid trafficking protect against strain injury. Interventions that impair deformation-induced lipid trafficking reduce the likelihood of plasma membrane resealing49,56 and increase cell death. It may help explain why the breaks seen across endothelial cells, after short periods of high transmural capillary pressure57 or overinflation4 are transient. In this latter study, rats were subjected to mechanical ventilation with 35 mm Hg PIP for only 2 minutes and allowed to recover for graded periods of up to 3 hours. The animals that were killed immediately after the challenge exhibited mild pulmonary edema with severe permeability alterations, as attested by increases in dry-lung weight and albumin space (Fig. 42-8). The very rapid occurrence of microvascular injury was ascertained by the ultrastructural study, which disclosed changes identical with those previously described after longer periods of ventilation.
Figure 42-8
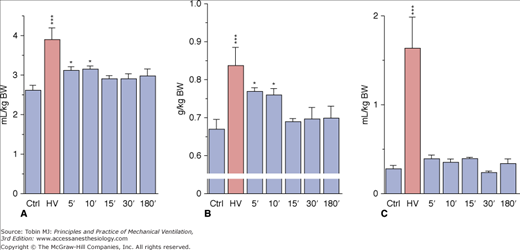
Effect of 45 cm H2O peak airway pressure ventilation for 2 minutes (HV) and followed by recovery for various lengths of time in intact rats. A. Extravascular lung water. B. Dry lung weight. C. Albumin distribution space. Permeability edema already present after 2 minutes cleared rapidly after cessation of ventilation. * = p < 0.05; *** = p < 0.001, as compared to controls. (Used, with permission, from Dreyfuss et al.4)
These data strongly suggest that, at least in small animals, vascular leakage after overinflation is almost immediate and extensive. During recovery, both extravascular lung water and dry lung weight promptly returned to normal (see Fig. 42-8A and B), indicating that resorption of edema can be very rapid. Edema resorption does not necessarily indicate restoration of the alveolo–capillary barrier. The 30-minute distribution space of 125I-albumin in lungs, however, was found to be normal, indicating that no albumin permeability defect was present after the time of tracer injection (see Fig. 42-8C). Because the tracer was injected during the recovery period (i.e., after cessation of overinflation), this finding demonstrated reversal of the alterations in permeability. ELF volume decreased after cessation of overinflation (see Fig. 42-3), reflecting resorption of the excess alveolar fluid concomitantly with the decrease in extravascular lung water. Even after 3 hours of recovery, however, ELF volume remained higher than in control animals. No marked changes in epithelial fluid protein content were observed (see Fig. 42-4B), reflecting the slower-than-water clearance of protein in alveolar edema fluid.58 The absence of notable protein resorption explains why the protein concentration in ELF increased during recovery (see Fig. 42-4A).
Ultrastructural Findings of Ventilator-Induced Lung Injury
Electron microscopic studies have confirmed that permeability alterations are prominent in the genesis of ventilator-associated pulmonary edema. Both endothelial and epithelial alterations have been observed.3,4,30,36
After short durations (5 to 10 minutes) of 45 cm H2O PIP mechanical ventilation in rats, striking capillary abnormalities consistent with pulmonary edema of the nonhydrostatic type were observed. Some endothelial cells were detached from their basement membrane, resulting in the formation of intracapillary blebs filled with plasma-like material (Fig. 42-9A). Endothelial cells exhibited focal disruptions (Fig. 42-9B). Bleb formation has been reported in experimental high-permeability edema, regardless of the nature of the causative agent, and in acute respiratory distress syndrome (ARDS),59–62 but not in experimental hydrostatic pulmonary edema.59,60,63 Longer durations (20 minutes) of high PIP ventilation in rats resulted in alveolar flooding and obvious permeability alterations. Pathologic studies showed that this severe edema was accompanied with diffuse damage to the alveolar–capillary barrier. In addition to the capillary lesions, ultrastructural studies disclosed profound alterations in the epithelial layer. The severity of alterations varied. In some sites, the epithelial lining appeared intact. In many areas, however, findings included discontinuities (Fig. 42-10A and B) and sometimes almost complete destruction of type I cells, leaving a denuded basement membrane (Fig. 42-10B and C). In contrast, type II cells appeared preserved. Hyaline membranes filled the alveolar spaces in many of the sections examined (Fig. 42-10C). Similar to the endothelial abnormalities, these lesions are not specific and occur in toxic injuries as well as in ARDS.59–62 In contrast, when pressures remain in the 30- to 40-mm Hg range, hydrostatic edema does not affect epithelial integrity.59,60,64
Figure 42-9
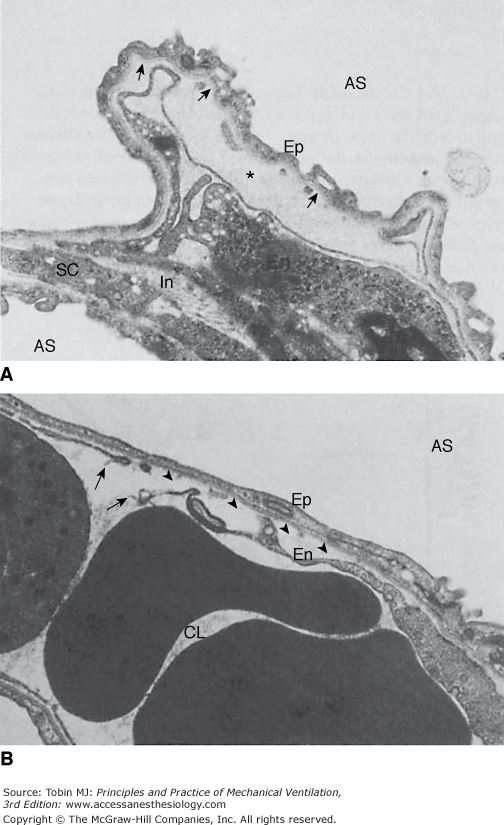
Ultrastructural features of the blood–air barrier after 5 minutes of 45 cm H2O peak airway pressure in a closed chest rat. A. The most striking change is the formation of an endothelial bleb secondary to detachment of the thin part of the endothelial cell (En) from the basement membrane (arrows). This bleb is filled with electron-dense material (*) of the same density as plasma. At this stage, epithelial type I cells (Ep) are intact. Note interstitial edema (In). AS, alveolar space; SC, septal cell. (Used, with permission, from Dreyfuss et al.3) B. Arrows indicate a disruption of the thin part of the endothelial cell detached from the basement membrane (arrowheads). CL, capillary lumen. (Courtesy Paul Soler, Ph.D.)
Figure 42-10
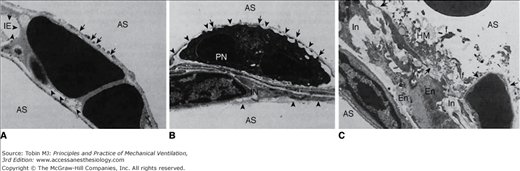
Ultrastructural features of the blood–air barrier after 20 minutes of 45 cm H2O peak airway pressure in a closed chest rat. A. Type I cells show numerous gaps (arrows). Arrowheads point at the basement membrane from which the endothelial cell is detached. AS, alveolar space; IE, interstitial edema. (Courtesy Dr. Paul Soler.) B. Epithelial and endothelial cells are almost completely destroyed, resulting in denuded basement membranes (arrowheads). A polymorphonuclear neutrophil (PN) inside the capillary lumen exhibits cytoplasmic processes, protruding through gaps in the capillary endothelium. IN, interstitium. (Courtesy Dr. Paul Soler.) C. Very severe alteration of the alveolar capillary barrier. Complete lysis of the epithelial layer (upper right quadrant) results in denudation of the basement membrane (arrows). Alveolar space is occupied by hyaline membranes (HM) composed of cell debris and fibrin (f). En, endothelial cells; In, interstitial edema. (Used, with permission, from Dreyfuss et al.3)
Studies using both transmission and scanning electron microscopy have demonstrated ultrastructural breaks in endothelial and epithelial cells when capillary transmural pressure was raised to 40 mm Hg or more.55,65,66 These breaks are the morphologic correlates of the increased microvascular permeability reported with very high microvascular pressures67,68 and described as the stretched-pore phenomenon.69,70 The similarities between capillary stress failure and VILI are quite remarkable. First, the electron microscopic appearance of endothelial and epithelial cell lesions exhibit similarities (Fig. 42-11).55,65 Second, both types of damage are partly reversible. Albumin leakage from capillaries during high-volume ventilation ceased almost immediately after discontinuation of ventilation.4 When capillary pressures were lowered to normal after elevation to a level causing stress failure, the number of endothelial and epithelial breaks fell compared with control experiments in which pressure remained elevated.57 More importantly, capillary stress failure is influenced by lung inflation: At a capillary transmural pressure of 32.5 cm H2O, increasing lung volume from a transpulmonary pressure of 5 to 20 cm H2O resulted in a significant increase in the number of capillary endothelium and alveolar epithelium breaks (Fig. 42-12).50 Thus, vascular pressures that are too low to affect microvascular permeability when lung volume is normal may produce permeability alterations when combined with a sufficiently marked increase in lung volume.
Figure 42-11

Examples of disruptions of the blood-gas barrier in in situ rabbit lungs perfused at 72.5 cm H2O capillary transmural pressure. A. Transmission electron microscopy. The alveolar epithelium and capillary endothelium are disrupted, but the basement membrane is intact. B. Scanning electron microscopy. Circular rupture involving only the epithelial layer (open arrow) and complete disruption of the blood-gas barrier (closed arrow) showing red blood cells in the opening (asterisk). A large amount of proteinaceous material is present in the alveolar lumen. The arrowhead indicates a type II cell with an intercellular junction (white arrow). (Used, with permission, from West et al.55 and Costello et al.66)
Figure 42-12
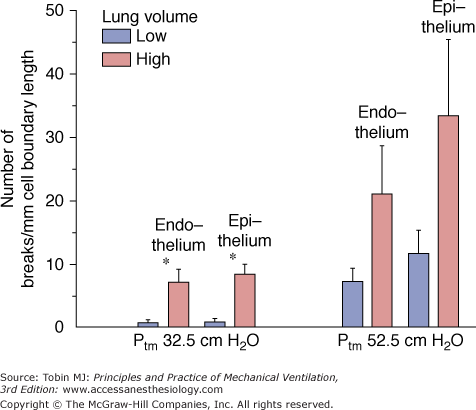
Effect of lung inflation on capillary stress failure. In situ rabbit lungs subjected to a capillary transmural pressure of 32.5 cm H2O had more endothelial and epithelial breaks at high lung volume (transpulmonary pressure of 20 cm H2O) than at low lung volume (transpulmonary pressure of 5 cm H2O). (Used, with permission, from Fu et al.50)
Effects of Activation of Inflammation by Protracted High-Volume Ventilation
The endothelial cell disruptions that have been observed during overinflation edema in small animals may allow direct contact between polymorphonuclear cells and basement membrane (see Fig. 42-10B) and promote leukocyte activation. Proinflammatory mediators may also be released by cells undergoing necrosis. Infiltration of inflammatory cells into the interstitial and alveolar spaces is not seen before several hours of injurious ventilation. In mice subjected to high PIP ventilation, leukocyte sequestration in lungs progressively increased with time.71 Woo and Hedley-White72 observed that overinflation produced edema in open-chest dogs, and that leukocytes accumulated in the vasculature and macrophages in the alveoli. Further studies confirmed these results73 and showed that high transpulmonary pressure increased the transit time of leukocytes in the lungs of rabbits.74 Conversely, high-volume pulmonary edema was less severe in neutrophil-depleted animals. Kawano et al75 observed that neutrophil-depleted rabbits had preserved gas exchange, little lung albumin leakage, and no hyaline membranes after 4 hours of mechanical ventilation in contrast to nondepleted animals in a saline-lavage model. Whereas it is predictable that healing of wounded tissue would involve inflammation, several authors have suggested that lung tissue stretching might result in lung damage solely through the release of inflammatory mediators and leukocyte recruitment, the so-called biotrauma hypothesis.76
The term biotrauma encompasses all molecular and cell-mediated mechanisms involved in VILI, such as the production of lung-borne cytokines.76,77 Although the causative role of those mediators on VILI has been debated,78 as comprehensively discussed in Chapter 42 of the second edition of this book and in “Pharmacologic Interventions” below, several investigators have tested the effect of cytokine modulators on lung dysfunction in experimental models of VILI.
Physiologic Determinants of Ventilator-Induced Injury
High inspiratory pressures consistently result in high lung volumes in normal animals. They induce well-documented hemodynamic changes in the systemic circulation,79 and may potentially affect blood-flow distribution in the lungs because parts of the lungs are placed under zone 1 condition over the ventilatory cycle. Another possibility is that high pressures per se produce regional distortions that may result in specific deleterious effects. Several studies have been conducted to determine the relative roles of intrathoracic pressure increases and lung distension in the genesis of pulmonary edema.
High-volume ventilation with low airway pressure was obtained by ventilating closed-chest rats with intermittent negative perithoracic pressures (using an iron lung). Ventilation with high airway pressures but low tidal volumes was obtained with thoracoabdominal strapping to limit thoracic movements. The effects of high PIP plus high tidal-volume ventilation were compared with those of intermittent negative airway pressure plus high tidal-volume ventilation and intermittently positive high PIP plus low tidal-volume ventilation.36 Pulmonary edema of the permeability type occurred in both groups of rats that received high tidal-volume ventilation, irrespective of inspiratory pressure (i.e., positive or negative; Fig. 42-13). In both groups, electron microscopic examination disclosed the same abnormalities as described above. In striking contrast with these findings, animals ventilated with a high PIP but a normal tidal volume had no edema (Fig. 42-13) and no ultrastructural changes. These findings have been replicated in rabbits80 and lambs.37
Figure 42-13
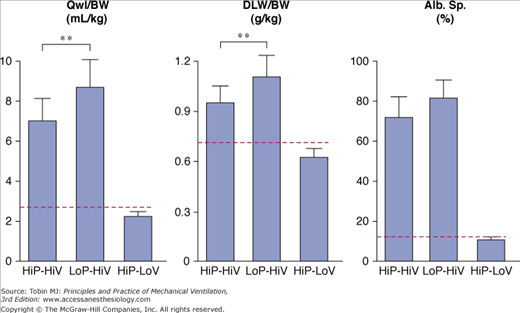
Comparison of the effects of high peak (45 cm H2O) positive inspiratory pressure plus high tidal-volume ventilation (HiP-HiV) with the effects of negative inspiratory airway pressure plus high tidal-volume ventilation (iron lung ventilation = LoP-HiV) and of high peak (45 cm H2O) positive inspiratory pressure plus low tidal-volume ventilation (thoracoabdominal strapping = HiP-LoV). Dotted lines represent the upper 95% confidence limit for control values. See Figure 42-6 for details on edema indexes. Permeability edema occurred in both groups receiving high tidal-volume ventilation. Animals ventilated with a high peak pressure and a normal tidal volume had no edema. ** = p < 0.01. (Used, with permission, from Dreyfuss et al.36)
A threshold volume for permeability changes as the lung is overexpanded was suggested by Carlton et al.37 These authors studied the effects of graded increases in tidal volume on lymph flow and protein concentrations in lambs. Peak inspiratory pressure (and therefore tidal volume) was increased in three successive steps (each lasting 4 hours) from baseline (16 cm H2O to 61 cm H2O). During the first two steps (33 and 43 cm H2O PIP), no changes in lymph flow or protein composition were observed. In contrast, when the highest PIP was reached (corresponding to a tidal volume of 57 mL/kg), lung lymph flow increased fourfold to sixfold compared with baseline. Similarly, the lymph-to-plasma-protein-concentration ratio did not change until the highest PIP was reached, at which time it increased significantly compared with controls. The investigators concluded that microvascular alterations in response to overinflation occurred beyond a pressure threshold rather than gradually as pressure increased. They pointed out, however, that the albumin-to-globulin ratio in lymph versus plasma decreased before maximum PIP was reached, suggesting altered protein sieving and probably abnormal microvascular permeability. Indeed, Tsuno et al39 demonstrated that ventilation of sheep with a moderately elevated PIP of 30 cm H2O (corresponding to a tidal volume of 30 mL/kg) for more than 40 hours invariably resulted in gross pathologic alterations and an increase in wet-lung weight. Ventilating healthy sheep with tidal volumes ranging from 9 to 51 mL/kg for 54 hours, Protti et al demonstrated that ventilator-induced lung edema developed only when a strain (i.e., the ratio between tidal volume and FRC81) greater than 1.5 to 2.0 was applied to the lungs.82 Interestingly, systemic inflammation and organ dysfunction also developed in pigs ventilated above this strain threshold.82 In smaller species, such as rats, in which VILI occurs more rapidly, edema was demonstrated with a PIP as low as 30 cm H2O, provided ventilation was sufficiently prolonged.2 Recently, scintigraphic methods allowing for real-time imaging of two-way protein fluxes across the alveolo-capillary barrier were used to measure simultaneous changes in alveolar and microvascular permeability during lung inflation in rats.51 The same end-inspiratory pressure threshold (between 20 and 25 cm H2O, corresponding to tidal volumes of 13.7 ± 4.69 and 22.2 ± 2.12 mL/kg) was observed for epithelial and endothelial permeability changes (Fig. 42-14). Of note, this threshold corresponds approximately to the pressure at which a decrease is observed in the slope of the respiratory system pressure–volume curve (the so-called upper inflection point) in rats.83
Figure 42-14
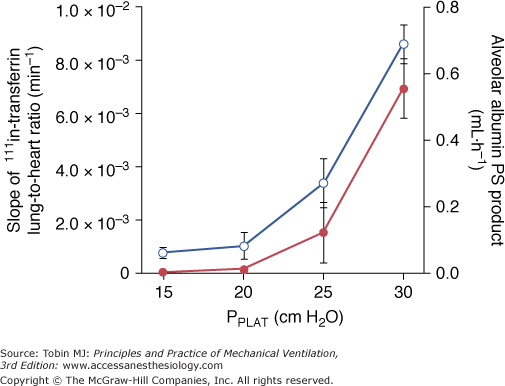
Relationship between plateau pressure (PPLAT) and 111In-transferrin lung-to-heart ratio slopes (left axis, open circles), reflecting lung-endothelial permeability, and alveolar 99mTc-albumin permeability-surface area product (right axis, full circles), reflecting lung-epithelial permeability. There is an increase in the permeability to proteins in both layers of the alveolo-capillary membrane above the same PPLAT threshold, between 20 and 25 cm H2O. (Used, with permission, from de Prost et al.51)
These studies emphasize that both the degree and the duration of lung overexpansion are crucial in determining the severity of pulmonary edema. Interestingly, the possibility of a potential pressure–volume threshold for additional lung injury during mechanical ventilation of patients with acute lung injury is also debated. This is discussed in “Clinical Relevance” below.
Respective Contributions of Sustained Inflation (Positive End-Expiratory Pressure) and Large Pressure–Volume Swings
For the same level of overall lung inflation, increasing FRC by PEEP results in less-severe alterations, the reasons for which may be reduction of VT at the same respiratory rate, and hence of peak inspiratory flow, or stabilization of terminal units.
Several studies assessing the deleterious effects of high gas flow rates on lung function, a minor determinant of VILI, are discussed in detail in Chapter 42 of the second edition of this book. Webb and Tierney showed that, for a given level of teleinspiratory pressure, edema was less severe when a 10-cm H2O PEEP was applied.2 It was subsequently shown36 that, although the amount of edema fluid was smaller with PEEP (Fig. 42-15), permeability alterations were similar to those observed with zero end-expiratory pressure. When rats were ventilated with PEEP, however, less edema occurred and, in particular, no alveolar flooding was observed on light microscopy.2,36 More strikingly, whereas diffuse alveolar damage was present in the animals ventilated with zero end-expiratory pressure, no epithelial-cell lining alterations were observed on electron microscopic examination of the lungs of animals ventilated with PEEP. The only ultrastructural alterations consisted of endothelial blebbing. It is worth noting that PEEP decreases lung-capillary blood volume,84 which, in turn, may lessen the capillary permeability alterations caused by high PIP ventilation.85
Figure 42-15
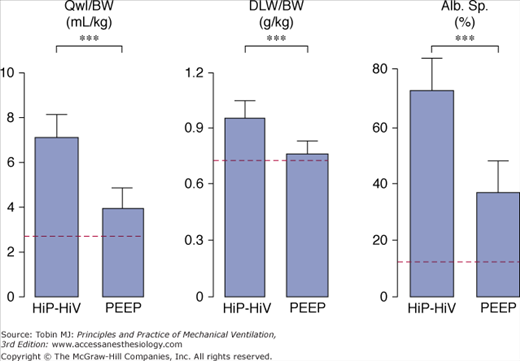
Effect of 10 cm H2O PEEP during high peak (45 cm H2O) positive inspiratory pressure plus high tidal-volume ventilation. Dotted lines represent the upper 95% confidence limits for control values. See Figure 42-6 for details on edema indexes. With PEEP, edema was less marked. ** = p < 0.001. (Used, with permission, from Dreyfuss et al.36)
This preservation of the alveolar–epithelial layer has received no satisfactory explanation. It may be that PEEP eliminated repetitive opening and closing of terminal airways, thereby decreasing shear stress and the development of surface-tension-induced epithelial cell damage at this level.86 Alternatively, avoidance of alveolar flooding by PEEP87,88 may have protected the epithelial lining from injury through the effects of yet unidentified humoral mediators.
One effect of PEEP is to reduce tidal volume for a given end-inspiratory pressure. It is interesting to note that studies using in situ perfused canine lobes89 found that, for equivalent perfusion-flow rates and microvascular hydrostatic pressures, the rate of hydrostatic edema formation increased with tidal volume. When an identical increase in mean airway pressure was achieved either by applying PEEP or by increasing tidal volume, edema was less marked under the former condition, suggesting that large cyclic changes in lung volume promote the development of edema. This explanation was also put forward by Corbridge et al,90 who observed in hydrochloric acid-injured dog lungs that ventilation with a large tidal volume and a low PEEP resulted in more severe edema than did ventilation with a small tidal volume and a high PEEP. The effect of the amplitude of tidal volume on alveolar epithelium protein permeability was confirmed in rats using noninvasive scintigraphic techniques.91 The alveolar albumin permeability-surface area product, measured from the clearance of an intratracheally instilled 99mTc-labeled albumin solution, dramatically increased, and in a dose-dependent manner, when VT was increased from 8 to 24 and 29 mL/kg.91
Finally, the potential role of hemodynamic alterations during PEEP ventilation should also be considered. For instance, rats ventilated with 45 cm H2O PIP and 10 cm H2O PEEP had more edema when PEEP-induced hemodynamic alterations were corrected by dopamine administration (Fig. 42-16).92 Moreover, arterial blood pressure was found to be significantly correlated with the amount of pulmonary edema under such conditions. The reason why use of PEEP during ventilation with high PIP is associated with reductions in both the amount of edema and the severity of cell damage may be a combination of hemodynamic alterations, shear stress reduction, and surfactant modifications. It should be borne in mind that this beneficial effect of PEEP during overinflation edema contrasts with the usual lack of reduction or even increase in edema reported with PEEP in most forms of experimental pulmonary edema.93,94
Figure 42-16
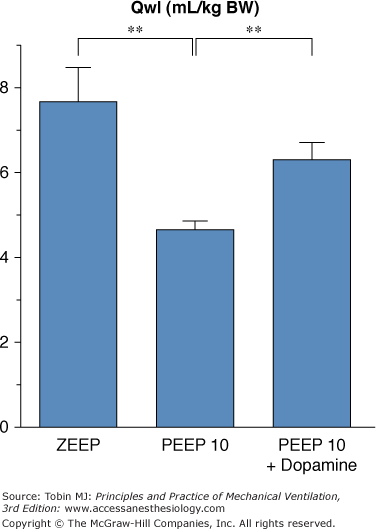
Effect of hemodynamic support with dopamine during 45 cm H2O peak pressure ventilation with 10 cm H2O PEEP on the amount of edema as evaluated by extravascular lung water. Compared with animals ventilated with 45 cm H2O peak pressure and zero end-expiratory pressure (ZEEP), animals ventilated with 45 cm H2O peak pressure ventilation with 10 cm H2O PEEP had less edema. This reduction of edema associated with PEEP was partly abolished when dopamine was administered. ** = p <.01. (Adapted, with permission, from Dreyfuss and Saumon.92)
It would be inappropriate to conclude from the data summarized above that tidal volume, which governs opening and closing of terminal units, is the sole determinant of VILI. On the contrary, the overall degree of lung distension (i.e., teleinspiratory volume) is probably the crucial factor. Hence, rats ventilated with a tidal volume within the physiologic range at two levels of PEEP (10 and 15 cm H2O) developed pulmonary edema only at the higher level of PEEP (Fig. 42-17).92 Similarly, doubling tidal volume had no effect in animals ventilated with zero end-expiratory pressure, but resulted in pulmonary edema when 10 cm H2O PEEP was used (Fig. 42-17).92 Thus, the safety of small tidal volumes depends on whether or not FRC is increased, and raising end-inspiratory volume by increasing FRC may cause lung injury independently of tidal volume.92
Figure 42-17
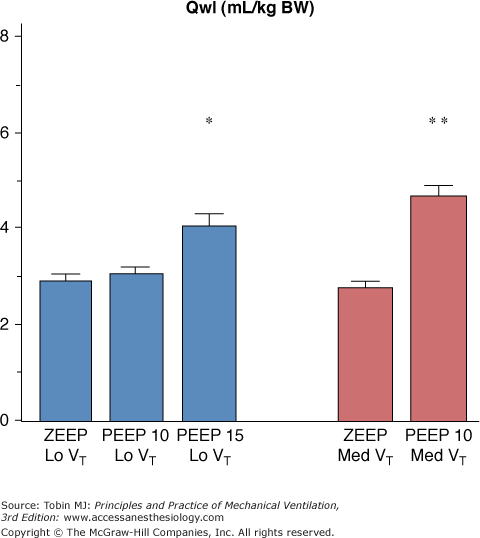
Effect of increasing PEEP from 0 to 15 cm H2O during ventilation with two levels of tidal volume (VT, 7 mL/kg of body weight [BW] = Lo VT; 14 mL/kg BW = Med VT). When PEEP was increased, pulmonary edema (as evaluated by extravascular lung water increases) occurred. The level of PEEP required to produce edema varied with tidal volume: 15 cm H2O PEEP during ventilation with a low tidal volume versus 10 cm H2O PEEP during ventilation with a moderately increased VT. * = p <.05; ** = p <.01 versus zero end-expiratory pressure (ZEEP) and the same VT. (Used, with permission, from Dreyfuss and Saumon.92)
It is now clear that VILI occurs whenever a certain degree of lung overinflation is reached, by whatever means. For a given level of teleinspiratory pressure (and volume), adjunction of PEEP seems to slow the development or diminish the severity of alterations95 but does not prevent the occurrence of permeability pulmonary edema.36,92
Effects of Ventilation on Previously Injured Lungs
This chapter does not consider the problems associated with lung prematurity, which is discussed in Chapter 23.
The aforementioned studies were conducted on animals with healthy lungs. It is conceivable, however, that diseased lungs may be more susceptible than healthy lungs to the deleterious effects of mechanical ventilation. Diseased lungs with a patchy distribution of lesions may be subjected to considerably greater regional stress than homogeneously inflated lungs. As stressed by Mead et al, the pressure tending to expand an atelectatic region surrounded by a fully expanded lung is approximately 140 cm H2O at a transpulmonary pressure of 30 cm H2O.96
Hernandez et al97 showed that whereas low doses of oleic acid or mechanical ventilation with PIP 25 cm H2O did not affect filtration coefficient and wet-to-dry ratio, the combination of the two did (Fig. 42-18). The same group also reported that the filtration coefficient increase observed during high PIP (30 to 45 cm H2O) ventilation of isolated perfused rabbit lungs was more marked following inactivation of surfactant by dioctyl succinate instillation.98 Moreover, whereas light microscopic examination showed only mild abnormalities (minimal hemorrhage and vascular congestion) in the animals subjected to ventilation only or surfactant inactivation only, the combination of both injuries caused severe damage (extensive hemorrhage, pulmonary edema, and hyaline membranes). Thus, ventilator-induced lung edema seems to develop at lower airway pressures in lungs with preexisting injury. Lung injury is usually inhomogeneous. The more compliant zones will thus receive the bulk of ventilation, which may favor their overinflation, a localized “baby lung effect.” Analysis of the pressure–volume curve may help understand how preexisting lung injury may interact with ventilator-induced injury. Before examining this interaction, it is important to understand how lung mechanical properties are modified by lung injury.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


