CHAPTER 24 TRAUMATIC BRAIN INJURY: PATHOPHYSIOLOGY, CLINICAL DIAGNOSIS, AND PREHOSPITAL AND EMERGENCY CENTER CARE
Death. Long-lasting or even permanent loss of function. Those are the burdens borne by many traumatic brain injury (TBI) patients and their families. Even some patients who initially appeared to have injuries that were mild according to clinical or radiographic criteria can suffer permanent injury.
INCIDENCE
According to data reported by the Centers for Disease Control and Prevention in the 1990s, 1.5 million Americans sustain a TBI every year. Of this number, hospitalization and ultimate survival occur in approximately 230,000 patients, but 50,000 will die. Long-term disability will occur in 80,000-90,000 patients annually. It has been estimated that more than 5 million men, women, and children in the United States are living with a permanent TBI-related disability.1
MECHANISM OF INJURY
Subdural Hematoma
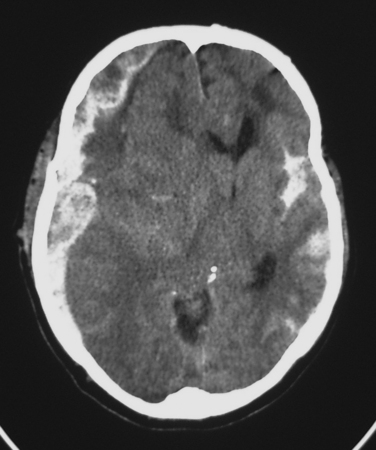
Classically, a subdural hematoma (SDH) (Figure 1) has been said to develop after tearing of a bridging vein, that is, a vein passing directly from the cortex to the overlying dura. The mechanical forces of the trauma can cause tearing of these veins. More recent evidence indicates that at least some of these hematomas actually form from splitting of inner and outer layers of the dura, that is, they may actually be “intradural hematomas.” Finally, some SDHs are caused by direct bleeding into the subdural space from parenchymal contusions or hematomas or from injured cortical arteries or veins.
Epidural Hematoma
Epidural hematomas (EDHs) classically arise after a blow to the side of the head results in a fracture of the thin temporal bone immediately overlying the middle meningeal artery. The patient may briefly lose consciousness after the initial impact, but he or she quickly awakens; thus, the brain injury was mild. Unfortunately, the fracturing of the skull lacerated the middle meningeal artery. Continued bleeding from this source produces an enlarging EDH, the presence of which may be signaled by such symptoms as severe and worsening headache, vomiting, and decreasing level of consciousness. The period between awakening from the initial concussion and subsequent lapsing into a coma has historically been described as a “lucid interval.” Importantly, loss of consciousness does not always occur after the skull is fractured, and many patients with large EDHs are awake until they begin to lapse into a terminal coma. It must also be mentioned that many EDHs are not associated with meningeal arterial bleeding. In these cases, perhaps the source of the hematoma is oozing from the overlying edges of fractured bone.
Subarachnoid Hemorrhage
The most common post-traumatic intracerebral hemorrhage is not a mass lesion, but rather diffuse subarachnoid hemorrhage (SAH) (see Figure 1). Several retrospective series report that SAH after TBI is independently associated with worse outcomes, but the mechanism that might explain such an association is unclear. In the acute setting, SAH does not seem to have much effect on patient management, which is driven instead by more immediately pressing concerns.
Ischemia
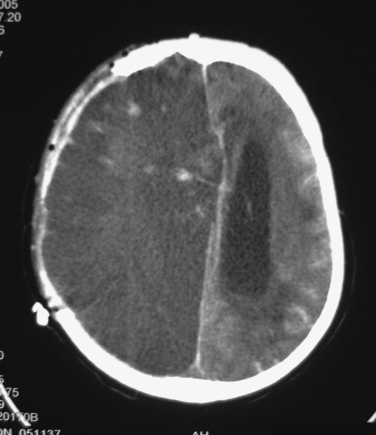
Diffuse injury may be of several types. Ischemia is a common form of diffuse injury. In some cases, mass effect from a traumatic hematoma may cause elevated intracranial pressure (ICP) and local compression of underlying tissue that can lead, respectively, to global and local reduction of cerebral blood flow (CBF). However, post-traumatic cerebral ischemia may also occur when no mass lesion is present, especially very early after injury. Although the CT scans in these cases may be relatively unimpressive, these patients may be quite vulnerable to the effects of such secondary insults as hypotension or hypoxia. Over the subsequent hours and days, CBF usually increases, but the damage from early ischemia may have already been done long before the increase in CBF (Figure 2). Some centers have used xenon CT or perfusion CT to measure CBF and identify ischemia immediately after injury.
Diffuse Axonal Injury
Another common type of diffuse injury is diffuse axonal injury (DAI). Although the axonal disconnection that characterizes DAI is commonly thought to occur at the time of injury, such immediate loss of axonal continuity probably occurs only when an injury produces severe cerebral parenchymal disruption. Instead, it appears more likely that the rotational and mechanical forces that are operant during the traumatic event produce a focal impairment of axoplasmic flow, which, in turn, culminates in axonal disconnection several hours after injury.2 This slight delay creates hope that a therapeutic window may exist for the administration of a yet-to-be-developed treatment that would prevent loss of axonal integrity and function.
CLINICAL DIAGNOSIS
Clinical Examination
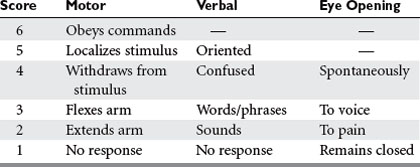
Many scales have been developed for the assessment of consciousness or neurologic status after injury, but by far the most widely used is the Glasgow Coma Scale (GCS)3 (Table 1). In conjunction with such information as the status of pupillary reactivity and the tempo or rate of change of a patient’s neurologic condition, the GCS is an extremely useful tool for assessing a patient’s baseline condition and subsequent progress.
< div class='tao-gold-member'>
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


