38 Traumatic Brain Injury
It is estimated that 3.2 million people are living with long-term disability related to traumatic brain injury (TBI).1 In addition to the personal toll, the direct and indirect costs of these disabilities are estimated to exceed $60 billion annually.2 Americans sustain an estimated 1.6 million TBIs each year. Approximately 290,000 require hospitalization, and 51,000 die of their injuries.3 However, the true incidence of TBI is unknown because current surveillance methodologies do not capture those treated in non-hospital settings (e.g., primary care office) or those who do not seek treatment at all.
TBI death rates in the United States fell during the 1980s. A substantial decline in motor vehicle–related fatalities was primarily responsible. At the same time, the incidence of gunshot wounds to the head rose, and in 1990, firearms surpassed motor vehicle crashes as the single largest cause of death due to TBI in some urban areas. However, in-hospital mortality rose to 8% during the last decade, presumably because of the increase in hospitalization rates of severe and moderate TBI.4
 Pathophysiology
Pathophysiology
Primary Injury
Injury to the brain is caused by external forces to the head that strain the tissue beyond its structural tolerance.5 These forces can be classified as contact or inertial.6 Contact forces typically produce focal injuries such as skull fractures, contusions, and epidural or subdural hematomas. Inertial forces result from the brain undergoing acceleration or deceleration (translational, rotational, or both) and can occur without head impact. Inertial forces can cause focal or diffuse brain injuries: pure translational acceleration leads to focal injuries such as contrecoup contusions, intracerebral hematomas, and subdural hematomas, whereas rotational or angular acceleration, common with high-speed motor vehicle crashes, usually causes diffuse injuries. Although external signs of head injury such as scalp abrasions, lacerations, and hematomas are common with blunt-force trauma, the brain can also be severely injured solely by inertial forces, without evidence of scalp or facial injuries.
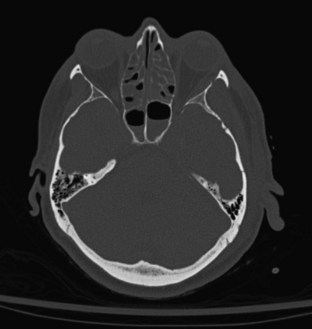
Skull fracture results from a contact force to the head that is usually severe enough to cause at least brief loss of consciousness. Linear fractures are the most common type of skull fracture and typically occur over the lateral convexities of the skull. Most often, they are nondisplaced cracks in the skull (linear fractures), but a particularly intense impact can cause a gap (diastasis) between the edges of the fracture. A depressed skull fracture, in which skull fragments are pushed into the cranial vault, usually results from blunt force by an object with a relatively small surface area, such as a hammer (Figure 38-1). The base of the skull can be fractured by severe blunt trauma to the forehead or the occiput. Basilar skull fractures are most common in the anterior skull base and often involve the cribriform plate, disrupting the olfactory nerves (Figure 38-2). Posterior basilar skull fractures may extend through the petrous bone and internal auditory canal, thereby damaging the acoustic and the facial nerves.
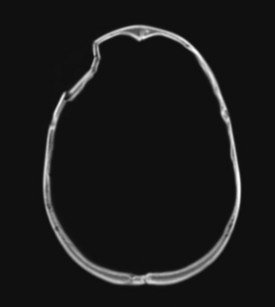
Figure 38-1 Right frontal depressed skull fracture caused by an assault with a hammer (axial CT scan, bone window).
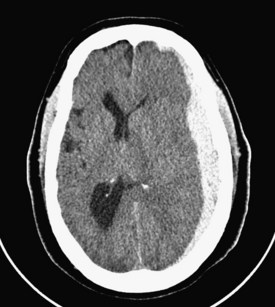
Common posttraumatic intracranial lesions are hemorrhage (epidural, subdural, and intraparenchymal), contusion, and diffuse brain injury. Subdural hematomas are seen in 20% to 25% of all comatose victims of TBI (Figure 38-3). They develop between the surface of the brain and the inner surface of the dura and are believed to result from the tearing of bridging veins over the cortical surface or from disruption of major venous sinuses or their tributaries. The hematoma typically spreads over most of the cerebral convexity; the dural reflections of the falx cerebri prevent expansion to the contralateral hemisphere. Swelling of the cerebral hemisphere is common in those with subdural hematomas, given the associated damage to underlying brain tissue. Underlying cerebral contusions were found in 67% of patients with subdural hematomas in one series.7 Subdural hematomas are classified as acute, subacute, or chronic, each having a characteristic appearance on computed tomography (CT): acute hematomas are bright white, subacute lesions are isodense with brain tissue and are therefore often overlooked, and chronic hematomas are hypodense relative to the brain.
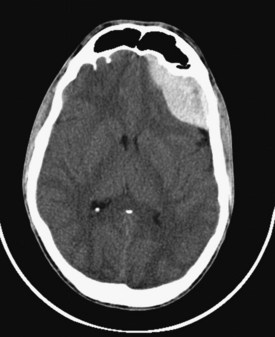
Epidural hematomas develop between the inner table of the skull and the dura, usually when the middle meningeal artery or one of its branches is torn by a skull fracture. They occur in 8% to 10% of those rendered comatose by TBI.8,9 The majority of epidural hematomas are located in the temporal or parietal regions, but they can also occur over the frontal or occipital lobes and (rarely) in the posterior fossa. They appear as hyperdense mass lesions on CT. Unlike subdural hematomas, their spread is limited by the suture lines of the skull, where the dura is very adherent. Because an epidural space normally does not exist, the clot must strip the dura from the inner table of the skull as it enlarges, resulting in its classic biconvex or lenticular shape (Figure 38-4). Epidural hematomas are uncommon in infants and toddlers, presumably because their skulls are more deformable and less likely to fracture, and in TBI victims older than 60 years, because the dura is extremely adherent to the skull.
Traumatic subarachnoid hemorrhage often results from tearing of the corticomeningeal vessels. Though common after severe TBI, subarachnoid hemorrhage does not produce a hematoma or mass effect.10 However, it may be associated with an increased risk for posttraumatic vasospasm, which may adversely affect clinical outcome.11
Contusions are heterogeneous lesions comprising punctate hemorrhage, edema, and necrosis and are often associated with other intracranial lesions. One or more contusions occur in 20% to 25% of patients with severe TBI. Because they evolve over time, contusions may not be evident on the initial CT scan or may appear as small areas of punctate hyperdensities (hemorrhages) with surrounding hypodensity (edema) (Figure 38-5). Local neuronal damage and hemorrhage lead to edema that may expand over the next 24 to 48 hours. With time, contusions may coalesce and look more like intracerebral hematomas. Depending on their size and location, they may cause significant mass effect, resulting in midline shift, subfalcine herniation, or transtentorial herniation. Contusions are most common in the inferior frontal cortex and the anterior temporal lobes,12 where the surface of the inner table of the skull is very irregular; they result from shifting of the brain over this irregular surface at the time of impact. Direct blunt-force trauma to the head can produce a contusion in the tissue underlying the point of impact (coup contusion). If the head was in motion upon collision with a rigid surface, a contusion may occur in the brain contralateral to the point of impact (contrecoup contusion).
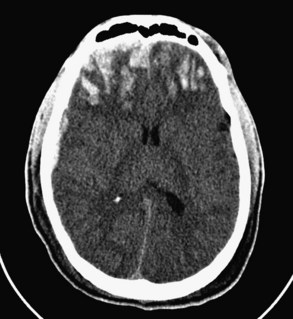
Diffuse axonal injury refers to lacerations or punctate contusions at the interface between the gray and white matter. Such punctate contusions are thought to result from the disparate densities of the gray and white matter and the consequent difference in centripetal force associated with a rotational vector of injury.13 Thus, diffuse axonal injury most often occurs after a high-speed motor vehicle crash, during which severe angular and rotational forces are applied to the head. Diffuse axonal injury was once thought to result solely from mechanical disruption at the time of impact; however, more recent research has identified cases in which the histologic footprints of diffuse axonal injury, such as fragmentation of axons and axonal swelling, do not appear until 24 to 48 hours after the incident, suggesting that some cases are a secondary manifestation of trauma.14,15 Diffuse axonal injury is present in almost half of all patients with severe TBI and in a third of those who die, and it is a common cause of persistent vegetative or minimally conscious state.
Posttraumatic intracranial lesions cause neurologic dysfunction via direct and in some cases indirect mechanisms. By destroying brain tissue, contusions and intraparenchymal hemorrhage cause deficits directly related to the function of the damaged tissue. Uncal herniation is also an important mechanism of temporary or permanent neurologic deficits.16,17 Semirigid dural reflections divide the intracranial contents into compartments. The tentorium cerebelli separates the anterior and middle cranial fossae from the posterior cranial fossa. The brainstem, specifically the midbrain, traverses an opening, the tentorial foramen, in the anterior central portion of this partition. The medial portion of the temporal lobe, the uncus, lies on both sides of the tentorial foramen. Because the most common TBIs, such as hematomas and contusions, are usually located over the lateral surfaces of the brain, and because the brain’s extreme lateral surface is the rigid skull, such lesions tend to depress the brain medially. Therefore, a subdural hematoma over the surface of the temporal lobe or a hemorrhagic contusion of the temporal lobe itself is likely to displace the medial portion of the temporal lobe (uncus) into the tentorial foramen (i.e., uncal herniation). Such displacement compresses the midbrain, which contains neurons that are part of the reticular activating system. At the base of the midbrain is the crus cerebri, which contains pyramidal fibers from the cortex, and the third cranial nerve, which exits the midbrain through the interpeduncular cistern. Midbrain compression due to uncal herniation damages the reticular activating system, causing loss of consciousness; stretches the third cranial nerve and its associated parasympathetic fibers, causing pupil dilatation and loss of the light reflex; and injures the pyramidal fibers in the crus cerebri, causing abnormal posturing responses in the contralateral arm and leg.
Intracranial hypertension is a major cause of posttraumatic neurologic morbidity and mortality.18 The intracranial pressure (ICP) is defined by the volume of CSF, blood, and brain tissue in the cranial vault. The volume of these components is dynamic, and the brain can accommodate moderate changes in any of the three. For example, the blood volume can rise or fall by as much as 30% to 40%, CSF absorption can increase to reduce the size of the ventricles by up to 90%, and brain tissue itself is compressible. Thus, the intracranial volume can gain 100 to 150 mL, equivalent to a moderate-sized subdural hematoma, without the ICP increasing significantly. When these buffering mechanisms have been exhausted, however, even a small increase in the size of a hematoma will cause a rapid rise in ICP. If appropriate treatment is delayed, the ICP may approach the mean arterial pressure (MAP), causing a hydrostatic block of blood flow to the brain and brain death. Intracranial hypertension, particularly if refractory to medical or surgical treatment, is the most common cause of death after severe TBI.
Secondary Injury
Posttraumatic ischemia initiates a cascade of metabolic events that lead to the surplus production of oxygen free radicals,19–21 excitatory amino acids,22,23 cytokines,24,25 and other inflammatory agents.26 Glutamate and aspartate are the excitatory amino acids most commonly implicated in excitotoxic injury,27 which is mediated by activation of N-methyl-D-aspartate, α-amino-3-hydroxy-5-methylisoazole-4-proprionic acid, or kainic acid receptors.23 Overactivation of these receptors causes an excessive influx of ionized calcium into the cytosol, and elevated amounts of ionized intracellular calcium play a key role in neurodegeneration after injury to the central nervous system (CNS).28,29 In addition, posttraumatic nonischemic events such as an increase in intracellular free Ca++ via receptor-gated or voltage-dependent ion channels induce the release of oxygen free radicals from mitochondria.30 Excessive levels of highly reactive oxygen free radicals cause lipid peroxidation of cell membranes, oxidation of intracellular proteins and nucleic acids, and activation of phospholipases A2 and C, which hydrolyze membrane phospholipids, thereby releasing arachidonic acid. The liberation of arachidonic acid triggers the generation of free fatty acids, leukotrienes, and thromboxane B2, all of which are associated with neurodegeneration and poor outcome after experimental TBI.31–33 Inflammatory cytokines, particularly interleukin (IL)-1, IL-6, and tumor necrosis factor, also are overproduced after TBI.34–36 In animal models, posttraumatic activation of microglia is a principal source of these cytokines.25 IL-1 and IL-6 provoke an exuberant cellular inflammatory response believed to be responsible for astrogliosis, edema, and tissue destruction.26,37
TBI also increases extracellular potassium levels,38 leading to an imbalance of intracellular and extracellular K+, disruption of the Na+/K+-ATPase cell membrane regulatory mechanisms, and subsequent cell swelling.39,40 Astrocyte swelling has been attributed to the clearance of excessive extracellular K+.41 High levels of extracellular K+ have also been implicated as the cause of widespread neuronal depolarization and spreading depression seen after experimental TBI.27,38,42 Moreover, potassium stimulates increased oxygen uptake in glial cells, potentially depriving adjacent neurons of oxygen.43,44 Severe TBI also causes a substantial decrease in extracellular magnesium (Mg++) levels, thereby impairing normal glycolysis, cellular respiration, oxidative phosphorylation, and the biosyntheses of DNA, RNA, and protein.45–47 Because Mg++ competes with Ca++ at voltage-gated cell membrane–associated Ca++ channels, reduced levels of Mg++ will result in an abnormal influx of Ca++ into the cell.
 Prehospital Care
Prehospital Care
The acutely injured brain is vulnerable to further damage from systemic hypotension, cerebral hypoperfusion, hypercarbia, hypoxemia, and elevated ICP. Preventing these physiologic insults is crucial to limiting secondary brain injury. Care of the TBI victim always should begin with evaluating and securing a patent airway and restoring normal breathing and circulation. Early endotracheal intubation usually benefits comatose patients. Securing and maintaining an airway are essential to optimal oxygenation and ventilation, and early intubation has been found to reduce mortality after severe TBI.48
The airway is usually most easily and safely secured by orotracheal intubation, a method in which most emergency medical personnel are trained and experienced. Patients with severe maxillofacial trauma may require nasotracheal intubation, but this is less desirable because it is a relatively blind procedure. The nasal passageways can be irritated, causing blood pressure (BP) and ICP to surge, and in those with severe anterior skull base fractures, the tube can inadvertently be passed into the brain. A third alternative for securing the airway is the laryngeal mask airway, an easily learned and rapidly applied device that has undergone successful field trials.49 However, it does not protect against aspiration and cannot be used to achieve high airway pressures. A surgical airway (cricothyroidotomy) should be performed only after other attempts to secure an airway have failed, and only by an experienced provider.
The patient should be sedated and pharmacologically paralyzed before intubation, because irritation of the oropharynx typically causes transient hypertension, tachycardia, increased ICP, and agitation that can interfere with the procedure. Fentanyl, a short-acting opioid agonist that produces analgesia and sedation, is the most commonly used sedative. The usual dose is 3 to 5 µg/kg body weight, administered intravenously (IV) 3 minutes before intubation. Etomidate, an alternative to opioids, provides adequate sedation and is less likely to cause hypotension. Some prefer thiopental because it is an ultra-short-acting barbiturate and is thus less likely to conceal the neurologic status when the patient reaches the trauma center; however, it is more likely than other agents to cause hypotension. Neuromuscular blocking agents commonly used for tracheal intubation include succinylcholine (1.5 mg/kg IV), which has the advantages of rapid onset, complete reliability, and very short duration of action. This last attribute is particularly important in the prehospital setting, where attempts at intubation sometimes fail. Vecuronium (0.01 mg/kg IV) is an alternative that offers the theoretical advantage of being a nondepolarizing muscle relaxant. Because it has a relatively long duration of action (1 to 2 hours), it is less forgiving of failed intubation attempts. Table 38-1 shows a recommended rapid-sequence intubation pathway.
TABLE 38-1 Recommended Rapid-Sequence Induction for Severely Head-Injured Patients
Rapid fluid resuscitation and restoration of a normal BP are critical in the prehospital setting, because hypotension has been associated with doubling of the mortality rate after severe TBI.50 The most likely cause of hypotension is hemorrhage, usually in the abdomen or chest; therefore, hypovolemia should be assumed. Lactated Ringer’s or normal saline solutions should be infused through a large-bore IV catheter as quickly as possible until normotension is achieved. Although preclinical studies suggest that hypertonic saline may be more effective than isotonic solutions for rapid volume resuscitation,51,52 results of several small clinical trials have not been convincing.53,54
In all cases of severe TBI, defined as a Glasgow Coma Scale (GCS) score of 3 to 8 and an inability to follow commands, patients should be treated as if they have a spinal fracture until an adequate examination of the spine proves otherwise. Among those who survive long enough to reach the emergency department, the likelihood of a cervical spine fracture is 2% to 7%. More troubling, however, is that an estimated 10% to 25% of all posttraumatic spinal cord injuries are iatrogenic, occurring during transport to the hospital.55 After respiratory and hemodynamic stabilization, the patient should be placed in a neutral position on a flat, hard surface. If the patient requires immediate tracheal intubation, it should be performed while another person provides in-line cervical spine immobilization. A rigid cervical spine collar should be placed as soon as possible. Next, the patient should be placed on a backboard; the cervical spine can then be further immobilized with a buttress of foam or towels placed on both sides of the head. To prevent any movement during transport, the patient should be strapped to the board in several locations.
The organization of emergency medical services and regional trauma programs has improved outcomes for victims of trauma, particularly those with severe TBI.56 A very large prospective study of the cost and outcomes associated with trauma center designation found more than a 25% reduction in in-hospital mortality for those with severe TBI who were initially treated at a level I trauma center compared to similarly injured patients treated at hospitals of similar size that were not designated trauma centers.57 This is likely because designation as a level I or II trauma center by the American College of Surgeons Committee on Trauma or a state health department ensures the availability of immediate neurosurgical care when the patient arrives. Therefore, every effort should be made to transport severely injured patients directly to a designated trauma center. Nonetheless, if an adequate airway or venous access cannot be obtained in the field, some patients may need to undergo respiratory or hemodynamic stabilization at a nearby emergency department en route to the trauma center. Once hemodynamic and airway stability is achieved, immediate transport to a designated trauma center should occur without delays for imaging or secondary surveys.
 Emergency Department Care
Emergency Department Care
Upon arrival at the trauma center, the emergency medical personnel should concisely report their prehospital assessment and management, including mechanism of injury, stabilizing maneuvers, medications given, initial vital signs, GCS score, and hemodynamic stability during transport. A thorough physical and radiographic examination to identify all life-threatening injuries should then be performed. Most trauma centers follow the Advanced Trauma Life Support protocol, a comprehensive routine that has proved successful in quickly detecting all major injuries.58 First the airway is reassessed, and the need for tracheal intubation is carefully reconsidered. For patients intubated in the field, proper placement of the tracheal tube is verified both clinically and radiographically. When the airway is secure and adequate oxygenation is confirmed using a percutaneous oxygen saturation monitor or arterial blood gas analysis, two large-bore IV catheters are inserted to provide sufficient venous access for high-volume fluid resuscitation. An isotonic saline solution is infused to continue volume replacement, which probably began at the scene. Any life-threatening injuries such as overt hemorrhage, tension pneumothorax, or cardiac tamponade should be treated immediately upon discovery. A brief neurologic examination is performed, including assessment of the GCS score (Table 38-2), pupillary size and reaction to light, and symmetry and extent of extremity movements. The head is palpated to detect fractures, lacerations, or penetrating wounds, and lacerations are probed gently to ascertain the presence of a depressed skull fracture or foreign body. Large lacerations are compressed with pressure dressings or temporarily sutured to prevent further hemorrhage. Careful inspection of the head should reveal hemotympanum, periorbital or mastoid ecchymosis, and CSF rhinorrhea or otorrhea.
TABLE 38-2 Glasgow Coma Scale59
| Response | Points |
|---|---|
| Speech | |
| Alert, oriented, and conversant | 5 |
| Confused, disoriented, but conversant | 4 |
| Intelligible words, not conversant | 3 |
| Unintelligible sounds | 2 |
| No verbalization, even with painful stimulus | 1 |
| Eye Opening | |
| Spontaneous | 4 |
| To verbal stimuli | 3 |
| To painful stimuli | 2 |
| None, even with painful stimuli | 1 |
| Motor | |
| Follows commands | 6 |
| Localizes painful stimulus | 5 |
| Withdraws from painful stimulus | 4 |
| Flexor posturing with central pain | 3 |
| Extensor posturing with central pain | 2 |
| No response to painful stimulus | 1 |
Data from Teasdale G, Jennett B. Assessment of coma and impaired consciousness: a practical scale. Lancet. 1974;2(7872):81-84.
Coagulopathy resulting from TBI is thought to occur when hypoperfusion causes activation of the protein C pathway, thereby inducing alterations in the clotting cascade.60 It is also commonly seen as a result of therapeutic anticoagulation with warfarin, especially in the geriatric population. Rapid identification and correction of coagulopathy is critical to prevent expansion of intracranial hematomas and allow surgical intervention. Fresh frozen plasma and cryoprecipitate can be used to correct the INR to 1.3 or less. Recombinant factor VIIa has been shown to decrease blood product requirements and costs associated with correction of coagulopathy.61 Other studies demonstrate more rapid correction times compared to blood product administration, although differences in outcomes have not been significant. Product expense and concern for thromboembolic complications have limited the routine use of recombinant factor VIIa.
 Definitive Treatment
Definitive Treatment
Critical to determining the severity of the brain injury and appropriate treatment are CT findings combined with a reliable postresuscitation GCS score and assessment of pupil size and reactivity. In the case of an acute subdural hematoma, for example, a patient with a moderate-sized lesion who has normal pupil size and reactivity and is able to follow commands might safely be treated nonoperatively. Conversely, surgery is unlikely to benefit an elderly patient with fixed and dilated pupils and a GCS score of 3 or 4, regardless of the CT findings. Other determining factors include size and location of the hematoma, presence and extent of an underlying contusion or brain swelling, and results of the neurologic examination. Neurologic deterioration, particularly a decline in mental status, suggests enlargement of the hematoma, and a new CT scan should be obtained promptly. Hematomas less than 10 mm thick that cause a midline shift of less than 5 mm can usually be observed, especially if they do not involve the middle cranial fossa.62 If nonoperative management is chosen for an intracranial hematoma, the patient should be monitored with frequent neurologic assessments in the intensive care unit (ICU). If the patient cannot follow commands, ICP monitoring is recommended.
The initial signs and symptoms of contusions vary greatly, depending on their size and location and the presence of other associated lesions. A small contusion may cause only a headache or no symptoms at all. If located in an eloquent area of the brain, such as the speech or motor areas, it may cause focal neurologic symptoms. Larger contusions, especially those involving the frontal or temporal lobes, typically cause elevated ICP and coma. Patients with small or deep-seated contusions without mass effect initially can be managed nonoperatively. The contusion should be followed closely with serial CT scans, however, because there is a 20% to 30% risk that the contusion will enlarge during the next 24 to 48 hours. The ICP should be monitored if the patient cannot follow commands. As with hematomas in the middle cranial fossa, contusions of the temporal lobes should be closely watched with CT scans. A temporal contusion can enlarge to the point of uncal herniation without a significant rise in ICP, so the threshold for evacuation of these lesions should be low (Figure 38-6). Unilateral frontal or temporal lobectomies are usually well tolerated and do not cause measurable neurologic deficits, while allowing space for further brain swelling.
< div class='tao-gold-member'>
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


