INTRODUCTION

AMPHETAMINE TOXICITY
Amphetamines as a class may be abused by ingestion, insufflation (“snorting”), parenteral injection, and smoking. “Ice” refers to a pure preparation of methamphetamine hydrochloride in a large crystalline form. Designer amphetamines include 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”), paramethoxyamphetamine (PMA), and synthetic cathinones (ingredients in “bath salts”). While the central nervous system (CNS) targets of these compounds are serotonergic and dopaminergic pathways, the clinical presentation is manifested as a sympathomimetic toxidrome.
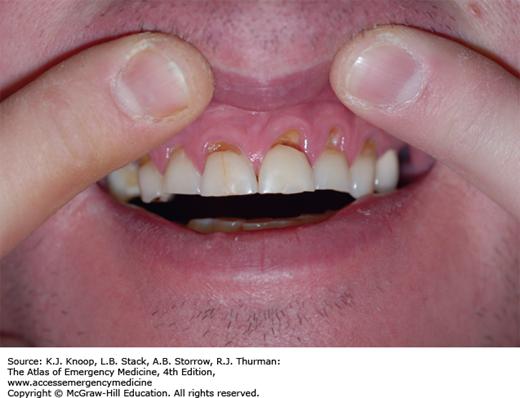
Although clinically indistinguishable from cocaine toxicity, the duration of effects is appreciably longer. The most common cardiovascular manifestations of toxicity are tachycardia and hypertension, although myocardial ischemia has been reported. Despite the cardiovascular effects, CNS toxicity is the primary reason most amphetamine users present for medical care. Presentations may range from increased anxiety to life-threatening agitated delirium with hyperthermia. Visual and tactile hallucinations and psychoses are common. Poor dentition is common among chronic users (“meth mouth”), and appears multifactorial in nature.
Treatment focuses upon the signs and symptoms of toxicity. As with other causes of sympathomimetic toxicity, initial management includes control of the agitation to prevent other complications (eg, rhabdomyolysis). Benzodiazepines are the first-line therapy for agitation; large repeated doses may be required. Severe hyperthermia requires evaporative cooling techniques. Hypertonic sodium may be useful for MDMA-associated cerebral edema and seizures.
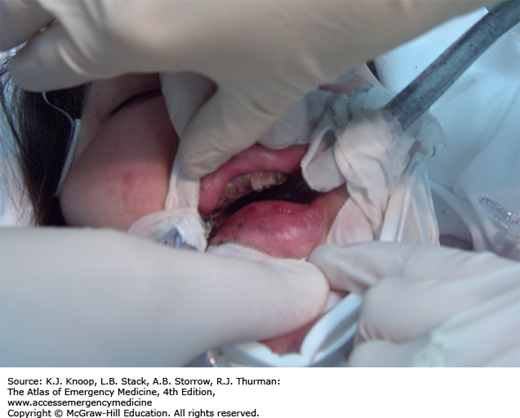
In addition to the medical complications associated with methamphetamine use, the manufacture of methamphetamine is associated with exposure to a number of toxic chemicals and risk for severe burns.
The hyperthermia associated with acute amphetamine poisoning may result in end-organ damage similar to patients with heat-stroke-like illness.
MDMA may result in syndrome of inappropriate antidiuretic hormone (SIADH) with subsequent hyponatremia and cerebral edema.

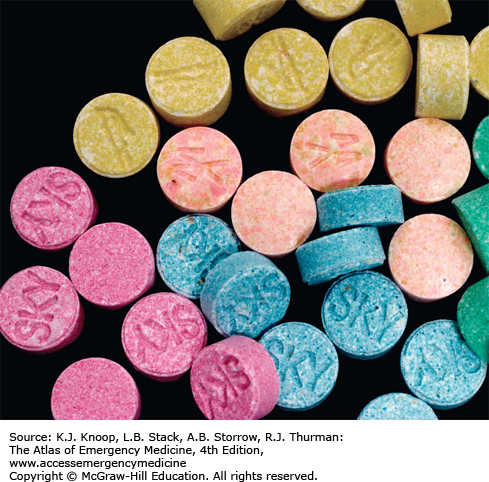
NEW DESIGNER DRUGS: “BATH SALTS” AND “SPICE”
The cathinones are derivatives of a naturally occurring phenethylamines found in the leaves of the Catha edulis (khat) plant. Some of these compounds include mephedrone, methylone, and methylenedioxypyrovalerone (MDPV). Adverse effects of these compounds include cardiac, psychiatric, and neurologic signs and symptoms, similar to effects seen with other sympathomimetics.
Synthetic cannabinoids have been promoted as “spice” or “incense” products that contain a substance usually labeled as “JWH” followed by a number. The designation reflects the original synthesis by John W. Huffman, an organic chemist at Clemson University. These compounds have full agonist effects at the cannabinoid receptors, in contrast with tetrahydrocannabinol (THC), which demonstrates only partial agonism. While cannabinoids such as marijuana do not typically result in sympathomimetic effects, the synthetic cannabinoids may also cause acute sympathomimetic toxicity including seizures and tachydysrhythmias.


Forensic analysis of these products has demonstrated significant variability in types and amounts of active products over time, both between and within marketed brands. As a result, individuals may experience different clinical effects with different exposures to the same product.
Patients under the influence of one of the synthetic compounds should be approached and managed similar to other acute sympathomimetic poisonings.
The ingredients in “bath salts” are not detected by routine testing for amphetamines as a class.
Agitation, psychosis, and seizures are common clinical effects of synthetic cannabinoid use.


COCAINE TOXICITY
Cocaine is a natural alkaloid derived from the leaves of Erythroxylum coca. Cocaine hydrochloride (powder cocaine) is a crystalline white powder. “Crack,” the free-base of cocaine hydrochloride, is an off-white substance named both for its rock-like appearance (“rock”) and due to the sound it makes when heated. In contrast to powder cocaine, crack may be smoked as it vaporizes instead of burning.
Cocaine intoxication manifests as a sympathomimetic toxidrome, with tachycardia, hypertension, diaphoresis, mydriasis, delirium, and hyperthermia. Increased muscular activity may result in rhabdomyolysis. Numerous neurological complications have been reported after cocaine use, including subarachnoid hemorrhage, intracerebral hemorrhage, cerebral infarction, and seizures.
Cardiovascular toxicity, including acute myocardial infarction, is well described after cocaine use. Dysrhythmias, including supraventricular tachycardia, atrial fibrillation and flutter, ventricular tachycardia, ventricular fibrillation, and torsades de pointes have been reported. Cocaine is a sodium channel blocker and may cause QRS widening on the electrocardiogram (ECG). Aortic dissection and rupture have been associated with cocaine use.
Pulmonary effects of cocaine use include reactive airway disease exacerbation, pneumothorax, pneumomediastinum, and cardiogenic and noncardiogenic pulmonary edema (NCPE). “Crack-lung” refers to an acute pulmonary syndrome of dyspnea, hypoxia, and diffuse pulmonary alveolar infiltrates.

Treatment is primarily supportive, and focuses upon the signs and symptoms of toxicity. No specific antidote exists. Cardiac monitoring is indicated for symptomatic patients. Initial management focuses upon control of agitation and hyperthermia, and prevention of complications (eg, rhabdomyolysis). The use of neuroleptic agents is relatively contraindicated for cocaine-associated psychomotor agitation due to the negative effects of these agents on thermoregulation, seizure threshold, and the potential for dysrhythmias. Benzodiazepines are the first line of therapy for agitation. They also appear beneficial in the management of cocaine-associated chest pain. Sodium bicarbonate administration should be considered for QRS widening in the setting of acute cocaine poisoning.
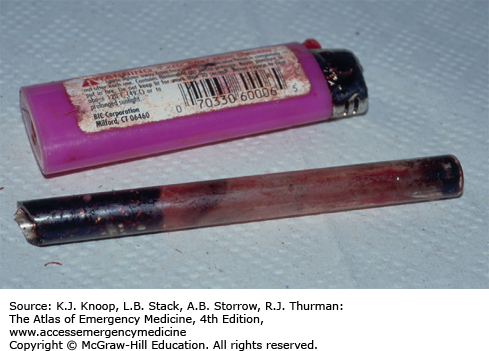
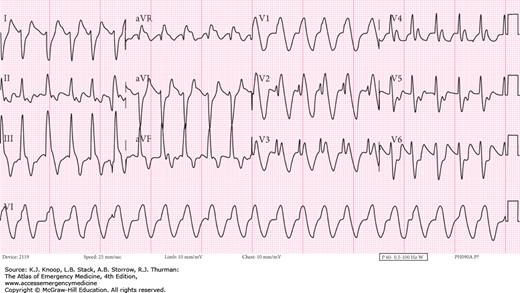
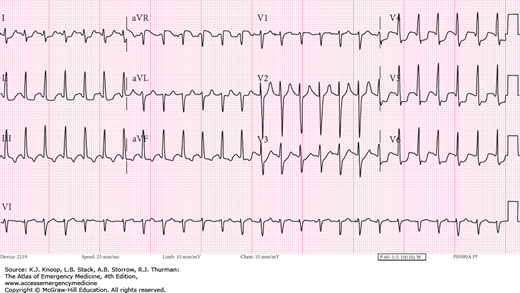
FIGURE 17.13
Treated Cocaine Cardiotoxicity. The 12-lead EKG of the same patient in Fig. 17.12, 68 minutes later after aggressive treatment with sodium bicarbonate to a serum pH of 7.26. (Photo contributors: Thomas Babcock, MD and Laurie Lawrence, MD.)
The use of β-blockers is contraindicated in the management of cocaine-associated chest pain and myocardial ischemia due to the potential for vasospasm and hypertensive crisis (“unopposed α-effect”).
Although the risk of myocardial infarction is greatest immediately after use, myocardial ischemia may occur up to 6 weeks after last use.
Cocaethylene may form in vivo after the use of cocaine and ethanol. Cocaethylene is more cardiotoxic than cocaine and has a longer half-life.
The rupture of a cocaine packet in a body-packer may result in fatal toxicity. Emergent surgical intervention may be considered for immediate removal of the packets.
ANTICHOLINERGIC (ANTIMUSCARINIC) TOXIDROME
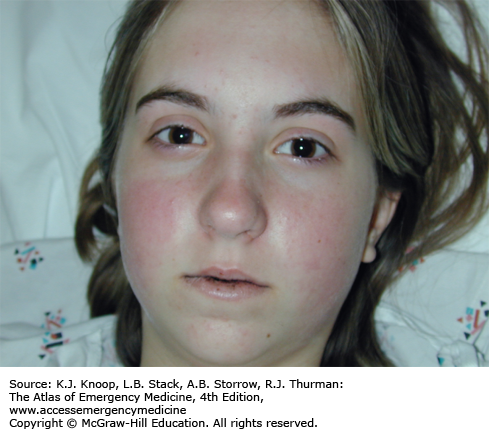
The anticholinergic toxidrome is well described by the mnemonic: hot as a hare, blind as a bat, mad as a hatter, red as a beet, and dry as a bone. As the etiology reflects central and peripheral muscarinic receptor blockade, it is more accurately termed an antimuscarinic toxidrome. A centrally mediated delirium may occur, which is typically not violent but is associated with mumbling speech and persistent “picking” behaviors. Other manifestations include hyperthermia, mydriasis, dry mucus membranes and axillae, tachycardia, decreased gastrointestinal (GI) motility, and urinary retention.
Many xenobiotics are antimuscarinic. One of the more common is diphenhydramine. Tricyclic antidepressants, (TCAs) phenothiazines, cyclobenzaprine, carbamazepine, atropine, scopolamine, glycopyrrolate, and belladonna alkaloids all have antimuscarinic properties. Plants such as jimson weed contain belladonna alkaloids and may be used recreationally.
Initial assessments of the vital signs and the duration of the QRS on ECG are critical. Since many antimuscarinic xenobiotics are also sodium channel blockers, QRS interval should be monitored. Hyperthermia occasionally occurs and is treated with evaporative cooling. Most of these patients require only supportive care, with the administration of benzodiazepines for agitation. A Foley catheter may be needed for treatment of the urinary retention. Occasionally, physostigmine is used as a diagnostic reversal agent for antimuscarinic poisoning, but its risks versus benefits must be considered. The half-life of physostigmine is only about 20 minutes.
The antihistamine diphenhydramine has sodium channel–blocking properties and may cause significant QRS widening.
Physostigmine crosses the blood-brain barrier, so an improvement in antimuscarinic delirium can be elicited with administration of the drug.
Glycopyrrolate is an antimuscarinic that does not cross the blood-brain barrier, so delirium does not occur.
Many features of anticholinergic toxidrome can mimic a sympathomimetic one—moist axillae on exam may help differentiate the two clinically as the anticholinergic toxidrome prevents sweating, while the sympathomimetic toxidrome promotes it.
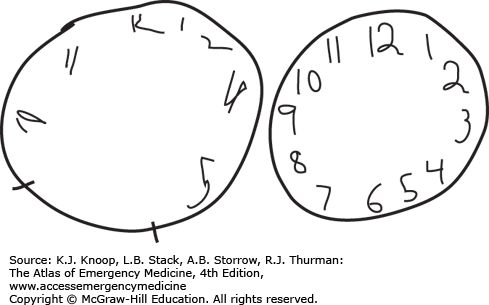
FIGURE 17.17
Anticholinergic Delirium. Prior to treatment with physostigmine, a patient suffering acute anticholinergic delirium drew the clock on the left. Following physostigmine administration, the patient drew the clock on the right. (Photo contributor: Division of Medical Toxicology, University of California, San Diego.)
CHOLINERGIC TOXIDROME
Acetylcholine (Ach) is a neurotransmitter of both the central and peripheral nervous systems and is the neurotransmitter for both muscarinic and nicotinic receptors. Inhibition of acetylcholinesterase increases Ach in the synapse. This results in a clinical syndrome producing both muscarinic and nicotinic effects. One mnemonic for the muscarinic effects is “DUMBBEL(L)S”: diarrhea, urination, miosis, bronchorrhea, bradycardia, emesis, lacrimation, salivation. Nicotinic effects may include fasciculations, paralysis, and occasionally mydriasis and tachycardia.
The most common cholinergic poisoning occurs after exposure to anticholinesterase insecticides. These include organic phosphorus (OP) compounds and carbamates. OP compounds bind to acetylcholinesterase, and after a period of time, this bond “ages” and becomes permanent. Carbamates are reversible binders of acetylcholinesterase and tend not to cross the blood-brain barrier.
The initial management of patients with acute cholinergic poisoning includes decontamination of clothing and skin. The airway should be secured early in the resuscitation as most patients have difficulty oxygenating and ventilating due to bronchorrhea and muscle weakness. Succinylcholine should be avoided as a paralytic agent in cholinergic crisis because succinylcholine requires acetylcholinesterase for its metabolism. Atropine is the initial therapeutic agent and dosing is doubled until the pulmonary secretions are dried. Pralidoxime is administered as early as possible for those patients with acute OP poisoning but would not be of benefit to those patients who have known carbamate poisoning. Seizures should be aggressively treated with benzodiazepines.
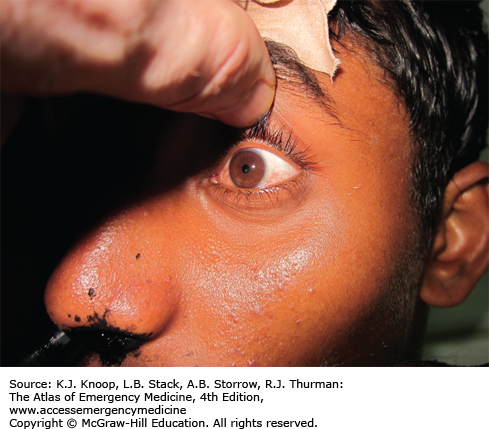
Patients developing central antimuscarinic toxicity during treatment with atropine who still require atropinization for secretion management may be treated with glycopyrrolate. Glycopyrrolate will not treat the central cholinergic effects as it does not cross the blood-brain barrier, but likewise will not cause central antimuscarinic toxicity.
The nicotinic clinical findings of cholinergic toxicity can be remembered by the days of the week: MTWtHF: mydriasis, tachycardia, weakness, hypertension, and fasciculations.
Nerve agents such as sarin, tabun, and soman are OP compounds and are readily absorbed through inhalational and dermal routes.
The pharmaceutical agents physostigmine, neostigmine, and pyridostigmine are carbamates; of these, only physostigmine crosses the blood-brain barrier.
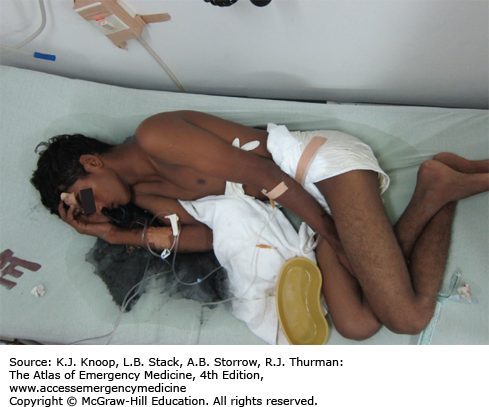
FIGURE 17.20
Atropine Therapy. This picture demonstrates the use of several vials of atropine that were required to dry the secretions of a patient with severe acute cholinergic toxicity. Multiple repeat atropine doses are commonly needed to treat cholinergic poisoning. (Photo contributor: Shannon Langston, MD.)

OPIOID TOXICITY
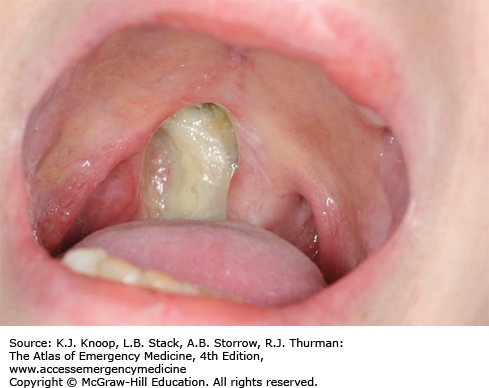
Opium is derived from the poppy plant, Papaver somniferum. Opiates are naturally occurring drugs derived from opium and include morphine, codeine, and paregoric. The term opioid refers to drugs with opium-like activity. Heroin is a bitter-tasting white powder; however, most street-grade heroin varies in color from white to dark brown. Mexican “black tar” heroin may be sticky like roofing tar or hard like coal, and appears dark brown to black in color. Diversion and abuse of legal narcotic agents is a common practice that is resulting in increased number of opioid deaths. For example, time-released preparations can be abused by chewing the tablets, snorting crushed tablets, or dissolving and parenterally administering the tablets, bypassing the sustained release mechanism. Significant mucosal necrosis may occur from the repeated snorting of crushed opioid tablets.
The classic opioid toxidrome is a clinical triad of coma, respiratory depression, and miosis. However, opioid-related CNS depression can range from mild sedation to coma. Normal or dilated pupils may occur after overdose of meperidine or pentazocine, or in the setting of CNS hypoxia. Death is typically due to respiratory depression. NCPE is associated with the use of certain opioids, including heroin, methadone, and morphine.
Care of these patients focuses upon airway management and antidotal therapy. Whole-bowel irrigation has been advocated after ingestion of sustained release formulations, or in the setting of body-packing and body-stuffing. In the latter, abdominal x-rays are indicated to look for evidence of foreign bodies. Chest radiographs are indicated for signs and symptoms of NCPE. Rhabdomyolysis and cerebral hypoxia may occur after prolonged periods of respiratory and CNS depression. Naloxone is the antidote of choice for significant opioid toxicity. In administering naloxone, care should be taken not to precipitate acute opioid withdrawal. Naloxone drips are indicated with longer acting opioids to avoid rebound respiratory depression from unopposed effects when the initial naloxone dose wears off.

FIGURE 17.22
Black Tar Heroin. Black tar heroin has a different appearance and texture than the South American and Asian heroin. Because it has a “gummier” texture, it is usually injected or smoked. Black tar heroin is associated with wound botulism. (Photo contributor: US Drug Enforcement Administration.)

FIGURE 17.23
Heroin-Related Noncardiogenic Pulmonary Edema. Noncardiogenic pulmonary edema may occur in the setting of opioid poisoning. The radiograph demonstrates the bilateral airspace opacities and the normal-sized cardiac silhouette. (Photo contributor: Division of Medical Toxicology, University of California, San Diego.)
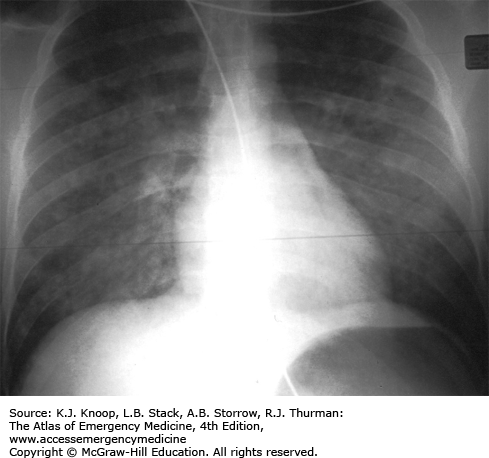
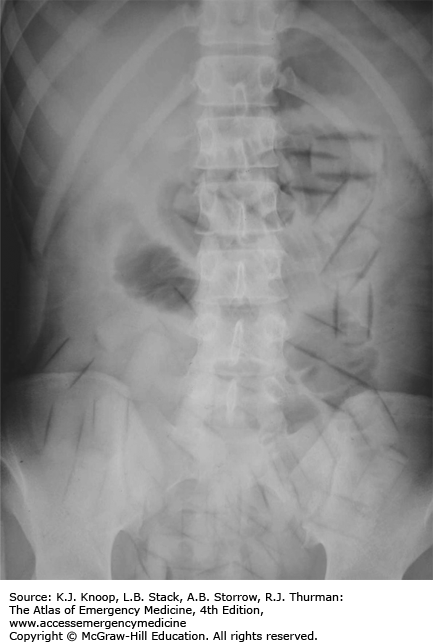
The presence of adulterants such as scopolamine or clenbuterol may mask or alter the appearance of the classic opioid toxidrome and may result in more significant toxicity than the primary drug.
Recurrent toxicity and life-threatening respiratory depression may occur following short-term reversal with naloxone administration, especially in body-stuffers or after ingestion of sustained release formulations.
The use of naloxone in the setting of tramadol toxicity is relatively contraindicated due to the occurrence of seizures.
The use of black tar heroin has been associated with wound botulism.
Poisoning from α2 agonists like clonidine mimics acute opioid poisoning.
ACETAMINOPHEN POISONING
Acetaminophen is a widely available analgesic and antipyretic agent. It is commonly found in combination with opioids, decongestants, antihistamines, and other over-the-counter and prescription products. Patients may complain of nausea and vomiting shortly after a toxic ingestion, but may also be asymptomatic. Signs and symptoms of acute liver injury typically occur within 36 hours after acute ingestion. Occasionally, patients present to the emergency department after developing evidence of hepatotoxicity, not realizing that the large ingestion of an acetaminophen-based product is the etiology.
In the overdose setting, acetaminophen exerts its toxic effects via a metabolite that is created via the P-450 enzyme system. The metabolite causes centrilobular necrosis of the liver which may lead to fulminant hepatic failure. Renal failure may also occur. Fatalities from hepatic failure usually occur 3 to 5 days after the ingestion. Treatment includes the administration of N-acetylcysteine (NAC), which can prevent acetaminophen-induced hepatotoxicity if initiated within 8 hours of the acute ingestion.

Activated charcoal may be considered in patients who present within 2 hours of acetaminophen overdose. A serum acetaminophen level drawn at 4 hours after a single acute ingestion can be plotted on the Rumack-Matthew nomogram to determine the need for treatment. If the serum level is at or above the treatment line, the patient should be treated with a standard course of oral or intravenously administered NAC. Patients who require administration of NAC should be admitted to the hospital.
Acetaminophen is a common agent in many over-the-counter medications. Patients who overdose on these medications require routine checking of a serum acetaminophen level to identify those who may need treatment with NAC.
The formulation of oral NAC is available in a 20% solution. The 20% solution comprises 20 g of NAC per 100 mL of solution. For the average 70-kg adult, the initial oral loading dose of 140 mg/kg would be 9.8 g, or approximately 50 mL of the 20% solution.
To enhance palatability, oral NAC can be diluted into a beverage of choice and served in a cup with a lid and straw.
Massive ingestions of acetaminophen may result in an anion gap metabolic acidosis.
SALICYLATE POISONING
Salicylates are a common cause of analgesic poisoning. Acute ingestions of large quantities of aspirin may have delayed absorption due to the formulation of the drug or the formation of bezoars. Poisoning may occur with chronic ingestions as well, particularly in older patients.
Early effects after ingestion include GI irritation which may lead to nausea and vomiting. Classically, salicylate-poisoned patients present with a mixed acid-base picture. Central stimulation of the respiratory drive results in a primary respiratory alkalosis. As a result of disrupted energy mechanics and decreased adenosine triphosphate (ATP) production, metabolic acidosis and lactate accumulation occur. The initial pH of the patient’s serum may be acidemic or alkalemic depending on the predominant acid-base disorder at the time of blood sampling. Ketonuria may also be noted. Hyperthermia occurs due to the generation and release of heat secondary to uncoupling of oxidative phosphorylation. Coma and seizures indicate severe nervous system toxicity and are associated with poor outcomes. Increased capillary permeability may result in NCPE and cerebral edema.
Fluid resuscitation to replace volume depletion is paramount early in the presentation. Since salicylate is a weak acid, alkalinizing the serum to a pH between 7.45 and 7.55 range traps the salicylate in an ionized form, decreasing entry into the CNS. Similarly, urinary alkalinization prevents tubular reabsorption, thereby enhancing the renal elimination of the salicylic acid. Because of the underlying metabolic acidosis and bicarbonate-induced hypokalemia, potassium replacement is usually needed in order to alkalinize the urine. Hemodialysis should be considered for deterioration in the acid-base status of the patient, renal failure, NCPE, or cerebral edema. A rapidly rising serum salicylate concentration is another consideration for dialysis. Patients with chronic ingestions may meet clinical criteria for extracorporeal elimination even with serum salicylate levels in the 40 to 50 mg/dL range. Admission should be strongly considered for any toxic salicylate ingestion.
Oil of wintergreen is typically 98% methylsalicylate (1400 mg/mL). One teaspoon (5 mL) provides the equivalent 7000 mg of salicylic acid.
If a patient with severe salicylism must be intubated, careful attention should be made to mimic the minute ventilation of the nonparalyzed patient. If a respiratory acidosis is allowed to occur, the patient will become severely acidemic which allows the salicylate to further distribute into the tissues and poison the mitochondria.
While acetazolamide administration results in alkalinization of the urine, the excretion of the bicarbonate into the urine comes at the expense of promoting acidemia, which could further drive the salicylate into the CNS and enhance toxicity.
The agent diflunisal will cause markedly elevated serum salicylate levels in the absence of other clinical or biochemical indicators of toxicity.
FIGURE 17.30
Aspirin Bezoar. Pill bezoar found in the GI tract of a patient who ingested approximately 750 enteric-coated aspirin tablets. At the time of death, approximately 13 hours after ingestion, the serum salicylate level was 128 mg/dL. More than 300 partially digested pills remained in the GI tract on postmortem. (Photo contributor: Jared M. Orrock, MD.)

FIGURE 17.31
Oil of Wintergreen. Severe salicylism may occur from ingestion of products that contain a high concentration of oil of wintergreen (methylsalicylate). This bottle of oil of wintergreen is a 98% solution, which contains the equivalent of 7000 mg of salicylate per teaspoon. (Photo contributor: R. Jason Thurman, MD.)
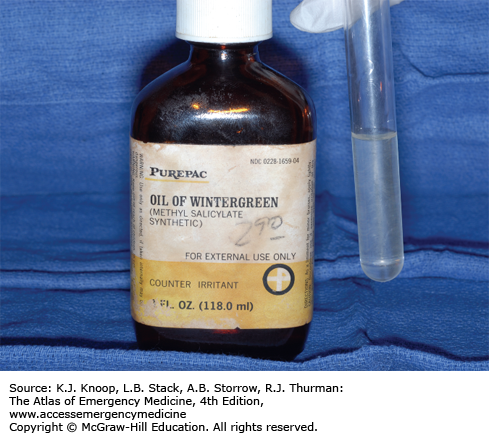
FIGURE 17.32
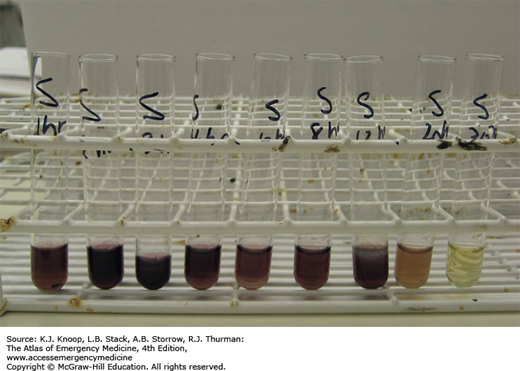
Trinder Reagent. In the presence of salicylates, the addition of Trinder reagent to urine specimen will yield a purple color. This picture demonstrates the reaction to Trinder reagent from urine samples collected serially 1 hour to 30 hours after ingestion of 650 mg aspirin. (Photo contributor: Sheila Dawling, PhD.)
TOXIC ALCOHOL INGESTION
The commonly available toxic alcohols include ethylene glycol, methanol, and isopropanol. Ethylene glycol is a sweet-tasting liquid commonly found in antifreeze, as well as in brake fluid. Methanol is used in lock deicers, windshield wiper fluid, and industrial solvents. Isopropanol is commonly marketed as “rubbing” alcohol, although it is also found in nonstreaking glass and window cleaners, soaps, cosmetics, and antifreezes.
The parent toxic alcohols have the potential to intoxicate, but are not otherwise toxic to end organs. Sequential metabolism via alcohol dehydrogenase and aldehyde dehydrogenase produces the organic acids responsible for end-organ toxicity and metabolic acidosis. Ethylene glycol is metabolized to glycolic and oxalic acids; the former is responsible for the acidosis, while the latter is responsible for calcium oxalate deposition in the renal tubules and delayed acute renal failure (24-72 hours postingestion). Hypocalcemia may occur with severe intoxication. Cranial nerve palsies may also occur 5 to 20 days after ingestion. Although less intoxicating than ethanol, methanol is metabolized to formic acid, which is responsible for both acidosis and direct retinal toxicity. Patients often report blurred or dim vision (“snowstorm”) prior to development of objective signs, including optic disc hyperemia, pupillary dilation, and poor accommodation. Pancreatitis and delayed basal ganglia lesions may occur. Isopropanol metabolism is limited to ketone formation and does not result in significant acidosis.
Decontamination options are limited, as activated charcoal does not bind alcohol and GI absorption is rapid. Emergency management is directed toward supportive care, diagnosis of the agent, and prevention of further metabolism. Methanol, ethylene glycol, and isopropanol levels may not be readily available. However, care should be taken in interpreting ancillary data, such as urinary fluorescence and the osmolar gap. Both ethanol and fomepizole competitively inhibit alcohol dehydrogenase. Both of these reduce metabolism of the parent compound, but do nothing for the toxic metabolites. Administration of folate (methanol) or pyridoxine and thiamine (ethylene glycol) may inhibit organic acid production or increase degradation by shunting metabolism to alternate pathways. Hemodialysis is indicated for signs of end-organ toxicity (eg, anion gap acidosis, renal failure, mental status changes) and should be considered for elevated toxic alcohol levels.
Provided adequate metabolism has occurred, isopropanol ingestion will demonstrate the presence of large amounts of ketones on a urine dipstick assay.
Only a few sips of concentrated methanol or ethylene glycol are required to produce toxicity in a toddler; these ingestions should be viewed as a “one pill can kill” exposure.
Coingestion of ethanol may delay development of eventual toxicity due to preferential blockade of alcohol dehydrogenase.
Both ethanol and fomepizole inhibit metabolism of the parent compounds.
Due to the occurrence of hypocalcemia in ethylene glycol poisoning, careful monitoring of ionized calcium is critical when using sodium bicarbonate in these patients.


FIGURE 17.36
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


