The Renal System and Anesthesia for Urologic Surgery
Mark Stafford-Smith
Andrew Shaw
Aaron Sandler
Catherine Kuhn
Key Points
Related Matter
Laparoscopic
Robotic Prostatectomy
Introduction and Context
The kidney plays a central role in implementing and controlling a variety of homeostatic functions, including excreting metabolic waste products in the urine, while keeping extracellular fluid volume and composition constant. Acute kidney injury (AKI) can occur as a direct result of surgical or medical disease, prolonged reduction in renal oxygen delivery, nephrotoxin insult, or a combination of these three factors. The first part of this chapter reviews renal physiology and pathophysiologic states as they relate to anesthetic practice, and then addresses strategies for recognizing and managing patients at risk for AKI and renal failure. The second part describes current urologic procedures and their attendant anesthetic management issues.
Renal Anatomy and Physiology
Gross Anatomy
The two normal kidneys are reddish-brown organs and are ovoid in outline, but the medial margin is deeply indented and concave at its middle (Fig. 49-1) where a wide, vertical cleft (the hilus) transmits structures entering and leaving the kidney. The hilus lies at approximately the level of the first lumbar vertebra. The kidneys lie in the paravertebral gutters, behind the peritoneum, with the right kidney lying slightly lower than the left one because of the presence of the liver. At its upper end, the ureter has dilated to give rise to the renal pelvis, which passes through the hilus into the kidney proper. There it is continuous with several short funnel-like tubes (calyces) that unite it with the renal parenchyma. The renal blood vessels lie anterior to the pelvis of the kidney, but some branches may pass posteriorly. Renal pain sensation is conveyed back to spinal cord segments T10 through L1 by sympathetic fibers. Sympathetic innervation is supplied by preganglionic fibers from T8 to L1. The vagus nerve provides parasympathetic innervation to the kidney, and the S2 to S4 spinal segments supply the ureters.
The kidneys are enclosed by a thick, fibrous capsule, itself surrounded by a fatty capsule that fills the space inside the loosely applied renal (Gerota’s) fascia. The developing kidney is first formed in the pelvis and then ascends to its final position on the posterior abdominal wall. During its ascent, the kidney receives blood supply from several successive sources, such that an accessory renal artery from the aorta may be found entering the lower pole of the kidney. When first formed, the rudimentary kidneys are close together and may fuse to give rise to a horseshoe kidney. This organ is unable to ascend, “held in place” by the inferior mesenteric artery, and thus when present it remains forever a pelvic organ.
The bladder is located in the retropubic space and receives its innervation from sympathetic nerves originating from T11 to L2, which conduct pain, touch, and temperature sensations, whereas bladder stretch sensation is transmitted via parasympathetic fibers from segments S2 to S4. Parasympathetics also provide the bladder with most of its motor innervation.
The prostate, penile urethra, and penis also receive sympathetic and parasympathetic fibers from the T11 to L2 and S2 to S4 segments, respectively. The pudendal nerve provides pain sensation to the penis via the dorsal nerve of the penis. Sensory innervation of the scrotum is via cutaneous nerves, which project to lumbosacral segments, whereas testicular sensation is conducted to lower thoracic and upper lumbar segments.
Ultrastructure
Inspection of the cut surface of the kidney reveals the paler cortex, adjacent to the capsule, and the darker, conical pyramids of the renal medulla (Fig. 49-1). The pyramids are radially striated and are covered with cortex, extending into the kidney as the renal columns. Collecting tubules from each lobe of the kidney (pyramid and its covering of cortex) discharge urine into the calyceal system via renal papillae at the entrance of each pyramid into the calyx proper. These collecting tubules originate deep within the radial striations (medullary rays) of the kidney and convey urine formed in the structural units of the kidneys, the nephrons. The parenchyma of each kidney contains approximately 1 × 106 tightly packed nephrons, each one consisting of a tuft of capillaries (the glomerulus) invaginated into the blind, expanded end (glomerular corpuscle) of a long tubule that leaves the renal corpuscle to form the proximal convoluted tubule in the cortex. This leads into the straight tubule, which loops down into the medullary pyramid (loop of Henle) and hence back to the cortex to become continuous with the distal convoluted tubule. This then opens into a collecting duct that is common to a number of nephrons and passes through the pyramid to enter the lesser calyx at the papilla. It is in
these parts of the nephron (proximal tubule, loop of Henle, distal tubule, and collecting duct) that urine is formed, concentrated, and conveyed to the ureters. The distal convoluted tubule comes into very close contact with the afferent glomerular arteriole, and the cells of each are there modified to form the juxtaglomerular apparatus, a complex physiologic feedback control mechanism contributing in part to the precise control of intra- and extrarenal hemodynamics that is a hallmark feature of the normally functioning kidney.
these parts of the nephron (proximal tubule, loop of Henle, distal tubule, and collecting duct) that urine is formed, concentrated, and conveyed to the ureters. The distal convoluted tubule comes into very close contact with the afferent glomerular arteriole, and the cells of each are there modified to form the juxtaglomerular apparatus, a complex physiologic feedback control mechanism contributing in part to the precise control of intra- and extrarenal hemodynamics that is a hallmark feature of the normally functioning kidney.
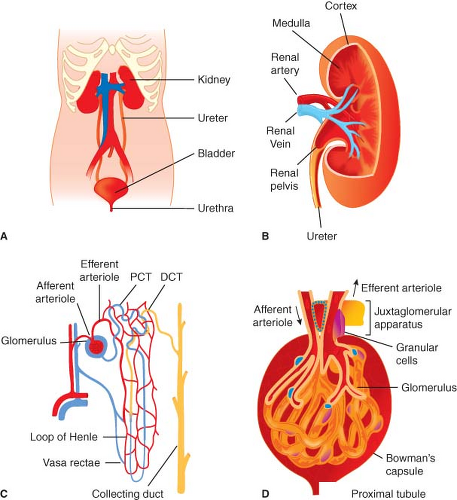 Figure 49.1. The gross anatomy (A) and internal structure of the genitourinary system and kidney. Internal organization of the kidney includes cortex and medulla regions and the vasculature (B). The nephron is the functional unit of the kidney (C). Plasma filtration occurs in the glomerulus (D); 20% of plasma that enters the glomerulus passes through the specialized capillary wall into the Bowman capsule and enters the tubule to be processed and generate urine. PCT, proximal convoluted tubule, DCT, distal convoluted tubule. (From: http://www.incontinenceaid.com/files/images/Urinary_system_components.jpg [A]; B; http://www.pathology.vcu.edu/education/PathLab/pages/renalpath/rpsrhome.htm from Lecture 1 (modified with permission) [C].) |
As is the case for the renal tubules, the vasculature of the kidney is highly organized. The renal artery enters the kidney at the hilum and then divides many times before producing the arcuate arteries that run along the boundary between cortex and outer medulla. Interlobular arteries branch from arcuate arteries toward the outer kidney surface, giving rise as they pass through the cortex to numerous afferent arterioles, each leading to a single glomerular capillary tuft. The barrier where filtration from the vascular to tubular space within the glomerulus occurs is highly specialized and includes fenestrated negatively charged capillary endothelial cells and tubular epithelial cells (podocytes) separated by a basement membrane. Normally, selective permeability permits approximately 25% of the plasma elements to pass into the Bowman capsule; only cells and proteins >60 to 70 kDa cannot cross. However, abnormalities of this barrier can occur with diseases that may permit filtration of much larger proteins and even red blood cells; these changes manifest as the nephrotic syndrome (proteinuria >3.5 g/24 hr) or glomerulonephritis (hematuria and proteinuria). The glomerular capillaries exit the Bowman capsule and merge to form the efferent arteriole and peritubular capillaries that nourish the tubules. The renal vasculature is unusual in having this arrangement of two capillary beds joined in series by arterioles. Blood supply to the entire tubular system comes from the glomerular efferent arteriole, which branches into an extensive capillary network. Some of these peritubular capillaries, the vasa recta, descend deep into the medulla to parallel the loops of Henle. The vasa recta then return in a cortical direction with the loops, join other peritubular capillaries, and empty into the cortical veins.
Correlation of Structure and Function
Because renal tissue makes up only 0.4% of body weight but receives 25% of cardiac output, the kidneys are by far the most highly perfused major organs in the body, and this facilitates plasma filtration at rates as high as 125 to 140 mL/min in adults. The functions of the kidney are many and varied, including waste filtration, endocrine and exocrine activities, immune and metabolic functions, and maintenance of physiologic homeostasis. As well as tight regulation of extracellular solutes such as sodium, potassium, hydrogen, bicarbonate, and glucose, the kidney also generates ammonia and glucose and eliminates nitrogenous and other metabolic wastes including urea, creatinine, and bilirubin. Finally, circulating hormones secreted by the kidney influences
red blood cell generation, calcium homeostasis, and systemic blood pressure.
red blood cell generation, calcium homeostasis, and systemic blood pressure.
The kidney fulfills its dual roles of waste excretion and body fluid management by filtering large amounts of fluid and solutes from the blood and secreting waste products into the tubular fluid. Filtration and reabsorption are affected by comorbid disease, surgery, and anesthesia and are the focus of the next section.
Glomerular Filtration
Production of urine begins with water and solute filtration from plasma flowing into the glomerulus via the afferent arteriole. The glomerular filtration rate (GFR) is a measure of glomerular function expressed as milliliters of plasma filtered per minute. The ultrafiltration constant (Kf) is directly related to glomerular capillary permeability and glomerular surface area. The two major determinants of filtration pressure are glomerular capillary pressure (PGC) and glomerular oncotic pressure (πGC). PGC is directly related to renal artery pressure and is heavily influenced by arteriolar tone at points upstream (afferent) and downstream (efferent) from the glomerulus. An increase in afferent arteriolar tone, as occurs with intense sympathetic or angiotensin II stimulation, causes filtration pressure and GFR to fall. Milder degrees of sympathetic or angiotensin activity cause a selective increase in efferent arteriolar tone, which tends to increase filtration pressure and GFR. The πGC is directly dependent on plasma oncotic pressure. Afferent arteriolar dilatation enhances GFR by increasing glomerular flow, which in turn elevates glomerular capillary pressure.
Autoregulation of Renal Blood Flow and Glomerular Filtration Rate
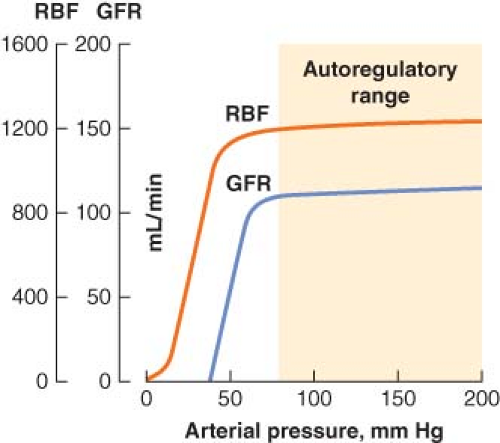 Figure 49.2. Renal blood flow (RBF) autoregulation maintains RBF and glomerular filtration rate (GFR) relatively constant with changes in systolic blood pressure from about 80 to 200 mm Hg. (From: http://www2.kumc.edu/ki/physiology/course/two/2_8.htm, with permission.) |
In health, autoregulation of RBF is effective over a wide range of systemic arterial pressures. Several mechanisms for regulating blood flow to the glomerulus have been described, and all involve modulation of afferent glomerular arteriolar tone. The myogenic reflex theory holds that an increase in arterial pressure causes the afferent arteriolar wall to stretch and then constrict (by reflex); likewise, a decrease in arterial pressure causes reflex afferent arteriolar dilatation. The other proposed mechanism of RBF autoregulation is a phenomenon called tubuloglomerular feedback, which is also responsible for autoregulation of GFR.
Tubuloglomerular feedback allows the composition of distal tubular fluid to influence glomerular function through actions involving the juxtaglomerular apparatus. When RBF falls, the concomitant decrease in GFR results in less chloride delivery to the juxtaglomerular apparatus, which causes the afferent arteriole to dilate. Glomerular flow and pressure then increase, and GFR returns to previous levels. Chloride also acts as the feedback signal for control of efferent arteriolar tone. When GFR falls, declining chloride delivery to the juxtaglomerular apparatus triggers release of renin, which ultimately causes the formation of angiotensin II. In response to angiotensin, efferent arteriolar constriction increases glomerular pressure, which increases glomerular filtration. It is important to realize that autoregulation of urine flow does not occur, and that above a mean arterial pressure of 50 mm Hg there is a linear relationship between mean arterial pressure and urine output.
Tubular Reabsorption of Sodium and Water
Reabsorption of water is a passive, osmotically driven process tied to the reabsorption of sodium and other solutes. Water reabsorption also depends on peritubular capillary pressure; high capillary pressure opposes water reabsorption and tends to increase urine output. The proximal tubule reabsorbs approximately 65% of filtered water in an isosmotic fashion with sodium and chloride. The descending limb of the loop of Henle allows water to follow osmotic gradients into the renal interstitium. However, the thin ascending limb and medullary thick ascending limb are relatively impermeable to water and play a key role in the production of concentrated urine. Only 15% of filtered water is reabsorbed by the loop of Henle; the remaining filtrate volume flows into the distal tubule. There, and in the collecting duct, water reabsorption is controlled entirely by antidiuretic hormone (ADH) secreted by the pituitary gland. Conservation of water and excretion of excess solute by the kidneys would be impossible without the ability to produce concentrated urine. This is accomplished by establishing a hyperosmotic medullary interstitium and regulation of water permeability of the distal tubule and collecting duct via the action of ADH.
gland releases ADH in response to an increase in either extracellular sodium concentration or extracellular osmolality. In addition, ADH release can be triggered by an absolute or relative reduction in intravascular fluid volume. The arterial baroreceptors are activated when hypovolemia leads to a decrease in blood pressure, whereas atrial receptors are stimulated by a decline in atrial filling pressure. Both of these circulatory reflex systems stimulate release of ADH from the pituitary and cause retention of water by the kidney in an effort to return the intravascular volume toward normal. ADH also causes renal cortical vasoconstriction when it is released in large amounts, such as during the physiologic stress response to trauma, surgery, or other critical illness. This induces a shift of RBF to the hypoxia-prone renal medulla.
The Renin–Angiotensin–Aldosterone System
Renin release by the afferent arteriole may be triggered by hypotension, increased tubular chloride concentration, or by sympathetic stimulation. Renin enhances angiotensin II production, which in turn induces renal efferent arteriolar vasoconstriction. Angiotensin II also promotes ADH release from the posterior pituitary, sodium reabsorption by the proximal tubule, and aldosterone release by the adrenal medulla. Aldosterone stimulates the distal tubule and collecting duct to reabsorb sodium (and water), resulting in intravascular volume expansion. Sympathetic nervous system stimulation may also directly cause release of aldosterone. This leads to renal cortical vasoconstriction, a decrease in GFR, and salt and water retention.
Renal Vasodilator Mechanisms
Opposing the saline retention and vasoconstriction observed in stress states are the actions of atrial natriuretic peptide (ANP), nitric oxide, and the renal prostaglandin system. ANP is released by the cardiac atria in response to increased stretch under conditions of volume expansion. Both natriuresis and aquaresis increase as ANP blocks reabsorption of sodium in the distal tubule and collecting duct. ANP also increases GFR, causes systemic vasodilatation, inhibits the release of renin, opposes production and action of angiotensin II, and decreases aldosterone secretion.1,2 Likewise, nitric oxide produced in the kidney opposes the renal vasoconstrictor effects of angiotensin II and the adrenergic nervous system, promotes sodium and water excretion, and participates in tubuloglomerular feedback.3
Clinical Assessment of the Kidney
Most agree that measures such as urine output correlate poorly with perioperative renal function5; however, much about the kidneys can be learned from knowing how effectively they clear circulating substances and inspection of the urine (i.e., urinalysis).
Renal Function Tests
Filtration is a useful method to clinically assess kidney function. As a key indicator of disease, knowledge of limited filtration capacity is important to guide drug dosing for agents cleared by the kidneys and helps with preoperative risk stratification. Also, acute declines in filtration capacity indicate kidney injury and predict a more complicated clinical course.2 GFR, as previously mentioned, refers to the plasma volume filtered per unit time by the kidneys, and normal values range from 90 to 140 mL/min. Normal GFRs relate to the patient age, size, and gender. In general, GFR declines 10% per decade after age 30 and is approximately 10 mL/min higher in men than women. A GFR below 60 mL/min meets criteria for chronic kidney disease (CKD) and is considered impaired, while values lower than 15 mL/min are often associated with uremic symptoms and may require dialysis.
An “ideal” substance to assess GFR through its clearance from the circulation must have specific properties, including a steady supply, free filtration, and no tubular reabsorption or excretion; ideally, it is also cheap and easy to measure. Unfortunately, the perfect ideal substance is yet to be identified. The “gold standard” GFR tools involve expensive and cumbersome measurements (e.g., inulin, 51Cr-EDTA or 99Tc-DTPA clearance), while the most practical and inexpensive test involves an imperfect “ideal” substance, creatinine. However, despite creatinine’s limitations, its relatively steady supply from muscle metabolism, modest tubular secretion, and proven usefulness in numerous clinical settings make it the most used renal filtration marker currently available. Although more ideal substances and other “early biomarkers” of AKI are being evaluated as clinical tools, current candidates (e.g., cystatin C) have yet to replace creatinine.
Estimates of GFR (eGFR) can be made by determining creatinine clearance (CrCl) from urine and blood creatinine tests. In stable, critically ill patients, 2-hour urine collections are sufficient to calculate CrCl,6 using the following formula:
where UCr = urine creatinine, V = total volume of urine collected, PCr = plasma creatinine, time = collection time.
However, if patient characteristics are known, GFR can also be estimated from a single steady-state serum creatinine value. Notably, predictive formulas are developed using data from stable (nonsurgical) populations, and factors such as fluid shifts, hemodilution, and hemorrhage may add an “unsteadiness” to perioperative eGFR using serum creatinine.
Nonetheless, serum creatinine remains, so far, an unsurpassed perioperative tool, particularly to reflect trends of change in renal filtration and to predict outcome, even during the perioperative period.7,8,9 Of the predictive formulas, the Cockroft–Gault equation is one of the oldest and most durable.10 The Cockroft–Gault equation uses patient gender, age (years), weight (kg), and serum creatinine (mg/dL):
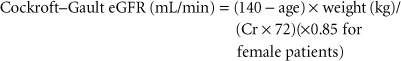
More recently, a method developed from the Modification of Diet in Renal Disease (MDRD) study that adds other factors including ethnicity (black vs. nonblack) to Cockroft–Gault equation has gained popularity.11
An abbreviated MDRD formula is available that can estimate GFR measured in milliliters per minute per 1.73 m2:

However, even a detailed MDRD eGFR under ideal conditions sometimes correlates poorly with a gold standard-determined GFR, with more than a 30% error in 10% of patients, and 2% deviating more than 50%.11
Some consensus definitions for significant perioperative renal dysfunction exist. For example, the Society of Thoracic Surgeons defines postoperative AKI as either a new requirement for dialysis or a rise in serum creatinine to >2 mg/dL involving at least a 50% increase in serum creatinine above baseline.12 Another definition requires a creatinine rise of >25% or 0.5 mg/dL (44 μmol/L) within 48 hours.13 The Acute Dialysis Quality Initiative (ADQI) Group definition for critically ill patients grades AKI by an acute creatinine rise of 50% as risk, 100% as injury, or 200% as failure (the RIFLE criteria).14 The Acute Kidney Injury Network (AKIN) definition, a 1.5-fold or 0.3 mg/dL (≥26.4 μmol/L) creatinine rise within a 48-hour period or more than 6 hours of oliguria (>0.5 mL/kg/hr), is a modification of its RIFLE predecessor.14,15 Notably, serum creatinine does not usually rise significantly until GFR rates fall below 50 mL/min, so preoperative serum creatinine may be normal in patients even with some degree of renal dysfunction (Fig. 49-3).
Blood urea nitrogen (BUN) is sometimes used to assess renal function but possesses few of the characteristics of an ideal substance. Tubular urea transport changes with some conditions (e.g., dehydration), and urea generation can be highly variable, particularly during the postoperative period (i.e., catabolic state). In addition, hemodilution (e.g., cardiopulmonary bypass [CPB]) may affect circulating BUN levels.
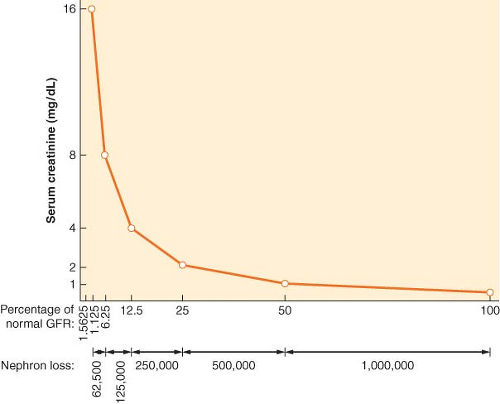 Figure 49.3. The nonlinear relationship between changes in renal filtration and serum creatinine level means that a large reduction (e.g., 75%, 120 to 30 mL/min) in glomerular filtration rate (GFR) may be associated with a modest rise in serum creatinine. Proportional reductions in GFR and (approximate) nephron loss (x axis) have an inverse logarithmic relationship with serum creatinine concentration (y axis). (Modified from: Faber MD, Kupin WL, Krishna G, et al. In: Lazarus JM, Brenner BM, eds. Acute Renal Failure: The differential diagnosis of ARF. 3rd ed. New York, NY: Churchill Livingstone, 1993, 133, with permission.) |
Urinalysis and Urine Characteristics
Urine inspection can reveal abnormal cloudiness, color, and unexpected odors. Detailed descriptions of urine examination are available16; therefore, only a summary is provided here. Cloudy urine is due to suspended elements such as white or red blood cells and/or crystals. Lightly centrifuged urine sediment will normally contain 80 ± 20 mg of protein per day and up to two red blood cells per high-power field (400×); higher levels of red blood cells or protein reflect abnormal kidney function. Urine protein electrophoresis can differentiate proteinuria from a glomerular (filtering), tubular (reuptake), overflow (supply that saturates the reuptake system), or tissue (e.g., kidney inflammation) abnormality.17 In contrast, color changes reflect dissolved substances; this occurs most commonly with dehydration, but other causes include food colorings, drugs, and liver disease (e.g., bilirubin). Unusual odors are less common but can also be diagnostic (e.g., maple syrup urine disease). Chromogenic “dipstick” chemical tests can determine urine pH and provide a semiquantitative analysis of protein, blood, nitrites, leukocyte esterase, glucose, ketones, urobilinogen, and bilirubin. In addition, microscopy can identify crystals, cells, tubular casts, and bacteria.
Urine specific gravity (the weight of urine relative to distilled water) normally ranges between 1.001 and 1.035, and can be used as a surrogate for osmolarity (normal 50 to 1,000 mOsm/kg), with 1.010 reflecting a specific gravity similar to that of plasma. High specific gravity (>1.018) implies preserved renal concentrating ability, unless high levels of glucose, protein, or contrast dye injection have raised specific gravity without significantly changing osmolarity.
Although poor urine output (e.g., <400 mL urine/24 hr) may reflect hypovolemia or impending prerenal renal failure, a majority of perioperative AKI episodes develop in the absence of oliguria.1 The normal response to hypovolemia is renal solute retention; fluid and electrolyte retention produces a concentrated urine with a low sodium content (<20 mEq/L). In contrast, impaired
concentrating ability due to AKI causes urine to approach plasma osmolarity (isosthenuria) with a higher sodium content (>40 mEq/L). The kidneys’ ability to retain electrolytes is also reflected in the fractional excretion of sodium (FENa), a test that uses a spot sample of urine and blood to compare sodium and creatinine excretion; this test can be useful to distinguish hypovolemia and renal injury:
concentrating ability due to AKI causes urine to approach plasma osmolarity (isosthenuria) with a higher sodium content (>40 mEq/L). The kidneys’ ability to retain electrolytes is also reflected in the fractional excretion of sodium (FENa), a test that uses a spot sample of urine and blood to compare sodium and creatinine excretion; this test can be useful to distinguish hypovolemia and renal injury:
where UNa = urine sodium, PNa = plasma sodium, UCr = urine creatinine, and PCr = plasma creatinine.
FENa <1% implies that sodium is being normally conserved while values above 1% are consistent with acute tubular necrosis (ATN).
Perioperative Nephrology
Pathophysiology
Altered renal function can be thought of as a clinical continuum ranging from the normal compensatory changes seen during stress to frank renal failure. Clinically, there is considerable overlap between compensated and decompensated renal dysfunctional states. The kidney under stress reacts in a predictable manner to help restore intravascular volume and maintain blood pressure. The sympathetic nervous system reacts to trauma, shock, or pain by releasing norepinephrine, which acts much like angiotensin II on the renal arterioles. Norepinephrine also activates the renin–angiotensin–aldosterone system and causes ADH release. The net result of modest activity of the stress response system is a shift of blood flow from the renal cortex to the medulla, avid sodium and water reabsorption, and decreased urine output. A more intense stress response may induce a decrease in RBF and GFR by causing afferent arteriolar constriction. If this extreme situation is not reversed, ischemic damage to the kidney may result, and AKI may become clinically manifest.
Electrolyte Disorders
Disorders of Sodium Balance
Hyponatremia is the most commonly occurring electrolyte disorder18,19 (see also Chapter 14). Symptoms rarely occur unless sodium values are <125 mmol/L, and these include a spectrum ranging from anorexia, nausea, and lethargy to convulsions, dysrhythmias, coma, and even death due to osmotic brain swelling.20,21,22 Hyponatremia may occur in the setting of an expanded (e.g., transurethral resection [TUR] syndrome), normal, or contracted extracellular fluid volume, and volemic status and urinary sodium concentration are key markers in differentiating the large number of potential causes of hyponatremia. If water excess is a reason for hyponatremia, dilute urine (sodium >20 mmol/L) is expected. Conversely, avid renal sodium retention (urine sodium <20 mmol/L) suggests sodium loss as a cause. If hyponatremia is acute, the risk of neurologic complications is higher, and cautious treatment is indicated to prevent cerebral edema and seizures. This should be accomplished with intravenous hypertonic saline and furosemide to enhance water excretion and prevent sodium overload (see transurethral resection syndrome section).
Hypernatremia (serum sodium >145 mmol/L) is generally the result of sodium gain or water loss, most commonly the latter. Dehydration of brain tissue can cause symptoms ranging from confusion to convulsions and coma. In cases of hypernatremia, laboratory studies often show evidence of hemoconcentration (increased hematocrit and serum protein concentrations). In addition, urine output is usually low (<500 mL/day) and hyperosmolar (>1,000 mOsm), with very low urinary sodium concentration and evidence of prerenal failure (elevations of BUN and serum creatinine). Occasionally, the urine is not maximally concentrated, suggesting an osmotic diuresis or an intrinsic renal disorder such as diabetes insipidus. The primary goal of treatment is restoration of serum tonicity, which can be achieved with isotonic or hypotonic parenteral fluids and/or diuretics unless irreversible renal injury is present, in which situation dialysis may be necessary.
Disorders of Potassium Balance
Even minor variations in serum potassium concentration can lead to symptoms such as skeletal muscle weakness, gastrointestinal ileus, myocardial depression, malignant ventricular dysrhythmias, and asystole. Nearly 98% of total body potassium is intracellular. Circulating potassium levels are tightly controlled via renal and gastrointestinal excretion and reabsorption, but potassium also moves between the intra- and extracellular compartments under the influence of insulin and β2-adrenoceptors. In the kidney, 70% of potassium reabsorption occurs in the proximal tubule and another 15% to 20% in the loop of Henle. The collecting duct is responsible for potassium excretion under the influence of aldosterone.
Hypokalemia may be due to a net potassium deficiency or transfer of extracellular potassium to the intracellular space. Notably, total body depletion may exist even with normal extracellular potassium levels (e.g., diabetic ketoacidosis). Causes of hypokalemia include extrarenal loss (e.g., vomiting, diarrhea), renal loss (impaired processing due to drugs, hormones, or inherited renal abnormalities), potassium shifts between the extra- and intracellular spaces (e.g., insulin therapy), and, occasionally, inadequate intake. Clinical manifestations of hypokalemia include ECG changes (flattened T waves—“no pot, no T”, U waves, prodysrhythmic state) and skeletal muscle weakness. Hypokalemia treatment involves supplementation with intravenous or oral potassium; however, overly rapid potassium intravenous administration should be avoided because it can cause hyperkalemic cardiac arrest.
If a patient has hyperkalemia (elevated serum potassium level >5.5 mEq/L), it is important to consider the duration of the condition as chronic hyperkalemia is far better tolerated than an acute rise. Other than laboratory artifacts (e.g., hemolyzed sample), causes of hyperkalemia include abnormal kidney excretion, abnormal cellular potassium release, or abnormal distribution between the intracellular and the extracellular space.
Disorders of Calcium, Magnesium, and Phosphorus
Most of a grown adult’s 1 to 2 kg of calcium is in bone (98%), with the remaining 2% in one of the three forms: Ionized, chelated, or protein-bound. Normal serum calcium values range between 8.5 and 10.2 mg/dL, but only the ionized fraction (50%) is biologically active and precisely regulated. Ionized extracellular calcium concentration (iCa++) is controlled by the combined actions of parathyroid hormone (PTH), calcitonin, and vitamin D and further modulated by dietary and environmental factors. Hypocalcemia due to reduced serum protein levels is physiologically unimportant. Extracellular magnesium represents only 0.3% of total (mainly intracellular) stores, making normal serum levels (1.6 to 2.2 mg/dL) a poor reflection of total body magnesium.
Phosphorus is a major intracellular anion that plays a role in regulation of glycolysis, ammoniagenesis, and calcium homeostasis and is an essential component of adenosine triphosphate and red blood cell 2,3-diphosphoglyceric acid synthesis.
Phosphorus is a major intracellular anion that plays a role in regulation of glycolysis, ammoniagenesis, and calcium homeostasis and is an essential component of adenosine triphosphate and red blood cell 2,3-diphosphoglyceric acid synthesis.
The clinical manifestations of hypocalcemia include cramping, digital numbness, laryngospasm, carpopedal spasm, bronchospasm, seizures, and respiratory arrest. A positive Chvostek sign (facial muscle twitching in response to tapping the facial nerve) or Trousseau sign (carpal spasm induced by brachial artery occlusion) are the classic hallmarks of hypocalcemia but in practice are often absent. Mental status changes, including irritability, depression, and impaired cognition, may also occur. Cardiac manifestations include QT interval shortening and dysrhythmias.
Hypocalcemia may be due to several mechanisms, including a decrease in PTH secretion or action, reduced vitamin D synthesis or action, resistance of bone to PTH or vitamin D effects, or calcium sequestration. Citrate used for regional anticoagulation with dialysis can also cause hypocalcemia and may also lead to hypomagnesemia from decreased PTH secretion. Parathyroidectomy during neck surgery reduces PTH levels and is a common cause of acquired hypoparathyroidism.
Clinical symptoms of hypercalcemia correlate with its acuity and include constipation, nausea and vomiting, drowsiness, lethargy, weakness, stupor, and coma. Cardiovascular manifestations may include hypertension, shortened QT interval, heart block, and other dysrhythmias. The most frequent causes of hypercalcemia are primary hyperparathyroidism and malignancy. Other causes include thiazide (increased renal calcium reabsorption) or lithium (inhibits PTH release) therapy and rarer medical conditions including granulomatous disease, thyrotoxicosis, and multiple endocrine neoplasia (MEN) types I and II.
Hypomagnesemia (<1.6 mg/dL) may sometimes be asymptomatic, but clinically important problems can and do manifest, including neuromuscular, cardiac, neurologic, and related electrolytic (hypokalemia and hypocalcemia) abnormalities. Causes of hypomagnesemia can be divided in four broad categories: Decreased intake, gastrointestinal loss, renal loss, and redistribution. Nutritional hypomagnesemia can result from malabsorption syndromes in patients receiving parenteral nutrition, and it is also present in 25% of alcoholics. Redistribution occurs with acute pancreatitis, administration of catecholamines, and in “hungry bone syndrome” after parathyroidectomy.10 Magnesium can be supplemented orally or via the parenteral route.
Clinical manifestations of hypermagnesemia (>4 to 6 mg/dL) are serious and potentially fatal. Minor symptoms include hypotension, nausea, vomiting, facial flushing, urinary retention, and ileus. In more extreme cases, flaccid skeletal muscular paralysis, hyporeflexia, bradycardia, bradydysrhythmias, respiratory depression, coma, and cardiac arrest may occur. Hypermagnesemia generally occurs in two clinical settings: Compromised renal function (GFR <20 mL/min) and excessive magnesium intake (e.g., excessive intravenous therapy in preeclampsia). Although mild hypermagnesemia in the setting of normal renal function can be treated with supportive care and withdrawal of the cause, in some cases dialysis is necessary.
Hypophosphatemia is clinically more important than hyperphosphatemia and can result in symptoms including muscle weakness, respiratory failure, and difficulty in weaning critically ill patients from mechanical ventilation when serum levels are <0.32 mmol/L. In addition, low phosphate levels may diminish oxygen delivery to tissues and rarely cause hemolysis. Hypophosphatemia can result from intracellular redistribution (from catecholamine therapy), from inadequate intake or absorption secondary to alcoholism or malnutrition, or from increased renal or gastrointestinal losses.12 Intravenous and oral supplementation can be used to treat hypophosphatemia.
Hyperphosphatemia (>5 mg/dL) is generally related to accompanying hypocalcemia although increased phosphate levels may also lead to calcium precipitation and decreased intestinal calcium absorption.13,14 Significantly elevated serum phosphate levels are most commonly due to reduced excretion from renal insufficiency but can also result from excess intake or redistribution of intracellular phosphorus. Treatment of chronic hyperphosphatemia includes dietary phosphate restriction and oral phosphate binders.
Acid–Base Disorders
The primary determinant of serum pH is the balance between plasma bicarbonate (HCO3−) concentration and the PCO2 in the extracellular space. Acid–base homeostasis involves tight regulation of HCO3− and PaCO2. Primary extracellular pH derangements due to abnormal bicarbonate reabsorption and proton (H+) elimination by the kidney lead to metabolic acidosis or alkalosis, while factors that abnormally affect respiratory drive influence PaCO2, leading to respiratory acidosis or alkalosis. Because combined problems are often seen in perioperative critically ill patients, an approach to both “pure” and “mixed” acid–base disorders is presented here.
Metabolic Acidosis
The anion gap (AG) represents the total serum concentration of unmeasured anions and can be calculated as AG = (Na+ + K+) – (HCO3− + Cl−). It allows differentiation of the causes of metabolic acidosis into normal AG (8 ± 4) and increased AG (>16 mmol/L) varieties. Conditions that cause an increase in negatively charged ions other than bicarbonate and chloride (e.g., lactate, salicylate) increase the AG. In contrast, non-AG metabolic acidosis results from renal or gastrointestinal HCO3− loss and is associated with high chloride levels (hyperchloremic metabolic acidosis). The usual compensatory response to all types of metabolic acidoses is hyperventilation, which leads to a partial pH correction toward normal. Winter’s formula predicts expected PaCO2 for a metabolic acidosis as follows: PaCO2 = (1.5 × HCO3−) + 8.
Metabolic Alkalosis
Metabolic alkalosis is a common primary acid–base disturbance associated with increased plasma HCO3−. Increased extracellular HCO3− is due to a net loss of H+ and/or addition of HCO3−. The most common cause of metabolic alkalosis is gastrointestinal acid loss due to vomiting or nasogastric suctioning; the resulting hypovolemia leads to secretion of renin and aldosterone and enhanced absorption of HCO3−. Thiazides and loop diuretics both induce a net loss of chloride and free water and can cause a volume “contraction” alkalosis.
Respiratory Acidosis
If the lungs fail to eliminate CO2, hypercapnia and respiratory acidosis result, characterized by increased PaCO2 and decreased blood pH. Acute and chronic causes can be differentiated by examining arterial pH, PaCO2, and HCO3− values. In the early phase of respiratory acidosis, increased PaCO2 stimulates renal generation and secretion of H+. The kidneys continue to adapt to the increased pH through greater titratable acid excretion (e.g., ammonium) and HCO3− generation. Therefore, acute respiratory
acidosis is characterized by an elevated PaCO2, acidemia, and a relatively normal HCO3−. In contrast, chronic respiratory acidosis is associated with an elevated HCO3− (often accompanied by a relatively normal pH) due to renal compensation.
acidosis is characterized by an elevated PaCO2, acidemia, and a relatively normal HCO3−. In contrast, chronic respiratory acidosis is associated with an elevated HCO3− (often accompanied by a relatively normal pH) due to renal compensation.
Respiratory Alkalosis
Increased minute ventilation is the primary cause of respiratory alkalosis, characterized by decreased PaCO2 and increased pH. Patients with acute, uncompensated respiratory alkalosis have normal plasma HCO3−. In chronic respiratory alkalosis, renal compensation leads to decreased plasma HCO3−. The causes of respiratory alkalosis relate to abnormal respiratory drive from stimulants or toxins (e.g., salicylate, caffeine, nicotine, progesterone), central nervous system abnormalities (e.g., anxiety, stroke, increased intracranial pressure), pulmonary abnormalities (e.g., pulmonary embolism, pneumonia), mechanical hyperventilation, or systemic conditions such as liver failure and sepsis.
Mixed Acid–Base Disorders
It is not uncommon for a metabolic derangement to coexist with a respiratory derangement, particularly in intensive care patients. A general approach to the diagnosis of mixed acid–base disorders requires a step-wise approach that begins with a focused history and physical examination. An arterial blood gas and a concurrent serum chemistry panel (including Na+, K+, Cl−, and total CO2 concentrations) should also be obtained, and the use of an acid–base map may help differentiate simple from mixed disorders (Fig. 49-4).
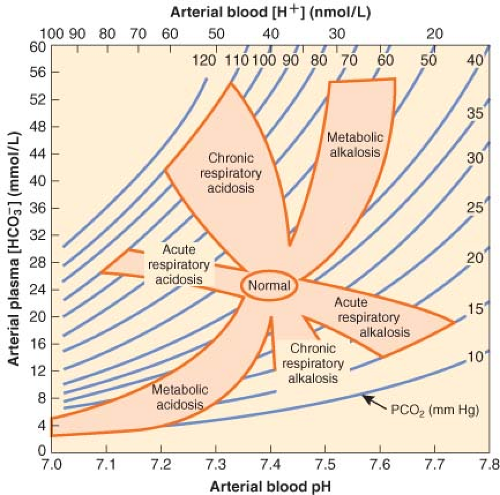 Figure 49.4. Acid–base map. Plotting the PCO2 and H+ (from the arterial blood gas) against plasma HCO3− (from the serum chemistry panel) for a patient can identify simple acid–base disorders. When mixed disorders exist, values may fall outside the shaded areas. (From: DuBose TD Jr. In: Brenner BM, ed. Acid-Base Disorders, Brenner & Rector’s The Kidney. 7th ed. Philadelphia, PA: WB Saunders; 2004:938, with permission.) |
Acute Kidney Conditions
Acute Kidney Injury
AKI can be caused by prerenal factors causing renal hypoperfusion, intrinsic renal causes, or postrenal causes (obstructive uropathy). There are many pathophysiologic similarities between the various causes of kidney injury.
Prerenal Azotemia
Prerenal azotemia is the increase in BUN associated with renal hypoperfusion or ischemia that has not yet caused renal parenchymal damage. The metabolically active cells of the medullary thick ascending limb of the loop of Henle are especially vulnerable to hypoxic damage because of their relatively high oxygen consumption.25 AKI ensues when necrosis of tubular cells releases debris into the tubules, causing flow obstruction, increased tubular back pressure, and leak of tubular fluid. Often, prerenal AKI is precipitated in patients with pre-existing renal vasoconstriction (e.g., volume depletion, heart failure, or sepsis) by nephrotoxin exposure or further reductions in cardiac output.26
Intrinsic Acute Kidney Injury
The term intrinsic not only implies a primary renal cause of AKI but also includes AKI due to ischemia, nephrotoxins, and renal parenchymal diseases. ATN remains the most common ischemic lesion and represents an extension of prerenal azotemia, whereas cortical necrosis may follow a massive renovascular insult such as prolonged suprarenal aortic clamping or renal artery embolism. Nephrotoxins often act in concert with hypoperfusion or underlying renal vasoconstrictive states to damage renal tubules or the microvasculature. Several common nephrotoxins, some of which are difficult to avoid in a hospitalized patient population, are listed in Table 49-1.
Postrenal Acute Kidney Injury (Obstructive Uropathy)
Downstream obstruction of the urinary collecting system is the least common pathway to established AKI, accounting for <5% of cases.27 Because it can generally be corrected, it is extremely important to exclude with a renal ultrasound examination as a source of AKI. The obstructing lesion may occur at any level of the collecting system, from the renal pelvis to the distal urethra. Intraluminal pressure rises and is eventually transmitted back to the glomerulus, thereby reducing glomerular filtration pressure and rate.
Table 49-1. Nephrotoxins Commonly Found in the Hospital Setting | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Nephrotoxins and Perioperative Acute Kidney Injury
Nephrotoxin exposure is a common occurrence in hospitalized patients and frequently plays a role in the cause of AKI in this population. Nephrotoxins may take the form of drugs, nontherapeutic chemicals, heavy metals, poisons, and endogenous compounds (Table 49-1). The nephrotoxins most likely to contribute to renal dysfunction/failure in the perioperative period are certain antimicrobial and chemotherapeutic–immunosuppressive agents, radiocontrast media, nonsteroidal anti-inflammatory drugs (NSAIDs), and the endogenous heme pigments myoglobin and hemoglobin. These diverse groups of renal toxins share a common pathophysiologic characteristic: They disturb either renal oxygen delivery or oxygen utilization and thereby promote renal ischemia.
Antimicrobial and chemotherapeutic–immunosuppressive agents are effective because they are cellular toxins. When these drugs are filtered, reabsorbed, secreted, and eventually excreted by the kidney, toxic concentrations in renal cells can be reached. The aminoglycoside antibiotics and amphotericin B are particularly difficult to avoid because they are effective antimicrobials, with few available alternatives. Their effect can be additive with other nephrotoxic factors causing impairment of kidney function. Hypovolemia, fever, renal vasoconstriction, and concomitant therapy with other nephrotoxic agents should be avoided wherever possible. Electrolyte disorders such as hypercalcemia, hypomagnesemia, hypokalemia, and metabolic acidosis can further enhance nephrotoxic damage to the kidney.
Cyclosporin A and tacrolimus are indispensable components of many immunosuppressive drug regimens, but in combination with other nephrotoxins and clinical factors, they can cause acute and exacerbate chronic kidney injuries in transplant recipients.23
Radiocontrast media poses a threat to the renal function of patients with diabetic nephropathy, pre-existing renal vasoconstriction (heart failure, hypovolemia), or renal insufficiency.24 Radiocontrast dye has effects on renal function that develop 24 to 48 hours after exposure and peak at 3 to 5 days. Measures that may prevent AKI or lessen the severity of renal damage include prehydration, smaller contrast doses, and judicious withholding of other nephrotoxins, such as NSAIDs. Elective surgery should be postponed until the effects of the dye have resolved. The idea that pretreatment with N-acetylcysteine can prevent radiocontrast nephropathy in patients with renal insufficiency25 has now largely been abandoned.
NSAIDs produce reversible inhibition of prostaglandin synthesis and are well-known nephrotoxins.26 Except in cases of massive overdose, NSAIDs produce renal dysfunction only in patients with coexisting renal hypoperfusion or vasoconstriction. Advanced age, hypovolemia, end-stage hepatic disease, heart failure, sepsis, chronic renal insufficiency, and major surgery are risk factors for development of NSAID-induced AKI.27
Myoglobin and hemoglobin are both capable of causing AKI in critically ill surgical patients. Myoglobin seems to be a more potent nephrotoxin than hemoglobin because it is more readily filtered at the glomerulus and can be reabsorbed by the renal tubules, where it chelates nitric oxide and thus induces medullary vasoconstriction and ischemia.28 Hypovolemia and acidemia potentiate the toxicity of both pigments. Reduced intravascular volume causes a decrease in RBF and GFR, which results in a smaller volume of tubular fluid with a relatively higher concentration of pigment. There is also evidence suggesting that pigment precipitation inside the tubular lumen is enhanced under acidotic conditions and that tubular obstruction plays a role in the pathogenesis of AKI.28,29
Preventive treatment of pigment-induced AKI is directed at increasing RBF and tubular (urine) flow while correcting any existing acidosis. These goals may be accomplished by expanding the intravascular fluid volume with crystalloid infusion, stimulating an osmotic diuresis with mannitol, and increasing the urine pH with intravenous bicarbonate therapy.30 Adequate systemic resuscitation from shock is a prerequisite if AKI is to be avoided, especially in massive crush injuries and electrical burns. Forced mannitol-alkali diuresis is indicated as the second step in the preventive treatment of myoglobinuria, with urine flow rates of up to 300 mL/hr and a urine pH of >6.5 advocated for patients with massive crush injuries.30
Chronic Kidney Disease
The uremic syndrome represents an extreme form of chronic renal failure, which occurs as the surviving nephron population and GFR decreases below 10% of normal. It results in inability of the kidney to perform its two major functions: Regulation of the volume and composition of the extracellular fluid and excretion of waste products. Water balance in ESRD becomes difficult to manage because the number of functioning nephrons is too small either to concentrate or to fully dilute the urine. This results in failure both to conserve water and to excrete excess water. Patients with uremic syndrome often require frequent or continuous dialysis.
Life-threatening hyperkalemia may occur in CKD because of slower-than-normal potassium clearance. Situations predisposing patients with renal failure to hyperkalemia are presented in Table 49-2. Derangements in calcium, magnesium, and phosphorus metabolism are also commonly seen in CKD (Table 49-3).
Table 49-2. Factors Contributing to Hyperkalemia in Chronic Renal Failure | ||
|---|---|---|
|
Table 49-3. The Uremic Syndrome | |
|---|---|
|
Metabolic acidosis occurs in two forms in ESRD: A hyperchloremic, normal AG acidosis and a high AG acidosis from inability to excrete titratable acids. Both render patients susceptible to an endogenous acid load such as may occur in shock states, hypovolemia, or with an increase in catabolism.
Cardiovascular complications of the uremic syndrome are primarily due to volume overload, high renin–angiotensin activity, autonomic nervous system hyperactivity, acidosis, and electrolyte disturbances. Hypertension due to extracellular fluid volume expansion, autonomic factors, and hyperreninemia is an almost universal finding in ESRD. Together with volume overload, acidemia, anemia, and possibly the presence of high-flow arteriovenous fistulae created for dialysis access, hypertension may contribute to the development of myocardial dysfunction and heart failure. Pericarditis may occur secondary to uremia or dialysis, with pericardial tamponade developing in 20% of the latter group.34 Pulmonary problems associated with CKD are limited to changes in lung water and control of ventilation. Pulmonary
edema and restrictive pulmonary dysfunction are commonly seen in patients with renal failure and are usually responsive to dialysis. Hypervolemia, heart failure, reduced serum oncotic pressure, and increased pulmonary capillary permeability are relevant factors in the development of pulmonary edema. Chronic metabolic acidosis may also be responsible for the hyperventilation seen in patients with ESRD, but increased lung water and poor pulmonary compliance can also stimulate hyperventilation.
edema and restrictive pulmonary dysfunction are commonly seen in patients with renal failure and are usually responsive to dialysis. Hypervolemia, heart failure, reduced serum oncotic pressure, and increased pulmonary capillary permeability are relevant factors in the development of pulmonary edema. Chronic metabolic acidosis may also be responsible for the hyperventilation seen in patients with ESRD, but increased lung water and poor pulmonary compliance can also stimulate hyperventilation.
The anemia of CKD occurs as a result of reduced levels of erythropoietin, red cell damage, ongoing gastrointestinal blood loss, and iron or vitamin deficiencies. Platelet dysfunction may aggravate blood loss, but it is responsive to dialysis, cryoprecipitate administration, and desmopressin acetate (or 1-deamino-8-D-arginine vasopressin [DDAVP]). Acquired defects in both cellular and humoral immunity probably account for the high prevalence of serious infections (60%) and high mortality from sepsis in CKD (30%).
Drug Prescribing in Renal Failure
If a drug depends solely on the kidney for clearance, then a simple approach to prescribing might involve a calculated percentage reduction in drug dosage that matches the reduction in GFR. Although GFR can be accurately measured, an estimated clearance derived from serum creatinine is usually adequate for these purposes. Unfortunately, clearance of most medications involves a more complex combination of both hepatic and renal functions, and drug level measurement or algorithms for specific drugs are often recommended.
AKI may affect absorption of a drug. For example, a reduced first-pass effect through the gastrointestinal tract and liver is associated with increased serum levels of oral β-blockers and opioids in patients with AKI. Also, an increase in the volume of distribution is seen in most patients with CKD due to increased plasma volume and decreased plasma protein binding. However, plasma protein binding is highly variable, with acidic drugs having reduced binding and basic agents (e.g., amide local anesthetics) having increased binding. Importantly, for drugs with less binding, “normal” drug levels may reflect dangerously high active (unbound) drug levels. For example, therapeutic phenytoin levels are typically reported as being in the range of 10 to 20 mg/mL normally but 4 to 10 mg/mL in cases of renal failure. Finally, hepatic metabolism of drugs is difficult to predict in the setting of renal failure because some hepatic enzymes are inhibited whereas others are induced, and accompanying liver disorders may alter the relationship of drug clearance with GFR.
Anesthetic Agents in Renal Failure
With the exception of methoxyflurane and possibly enflurane, anesthetic agents do not directly cause renal dysfunction or interfere with the normal compensatory mechanisms activated by the stress response. The nephrotoxicity of methoxyflurane appears to be due to its metabolism, which results in release of the fluoride ions believed responsible for the renal injury.35 It has been suggested that renal, not hepatic, metabolism of methoxyflurane may be responsible for generating fluoride ions locally that contribute to nephrotoxicity.36 Enflurane nephrotoxicity may also occur37 but is of minor clinical importance, even in patients with pre-existing renal dysfunction. Although direct anesthetic effects on the kidney are usually not harmful, indirect effects may combine with hypovolemia, shock, nephrotoxin exposure, or other renal vasoconstrictive states to produce renal dysfunction. If the chosen anesthetic technique causes a protracted reduction in cardiac output or sustained hypotension that coincides with a period of intense renal vasoconstriction, renal dysfunction or failure could result. This is true for either general or regional anesthesia. There are no comparative studies demonstrating superior renal protection or improved renal outcome with general versus regional anesthesia.
Significant renal impairment may affect the disposition, metabolism, and excretion of the commonly used anesthetic agents. Inhalation anesthetics are, of course, an exception to the rule that drugs with central nervous system activity (which generally are lipid soluble) must be converted to more hydrophilic compounds by the liver before being excreted by the kidney. The water-soluble metabolites of agents that are not inhaled may accumulate in renal failure and display prolonged pharmacodynamic effects if they possess even a small percentage of the pharmacologic activity of the parent drug. Drugs that are eliminated unchanged by the kidneys (e.g., certain nondepolarizing muscle relaxants, the cholinesterase inhibitors, many antibiotics, digoxin) have a prolonged elimination half-life when given to patients with kidney failure. Many drugs used in anesthesia are highly protein-bound and may demonstrate exaggerated clinical effects when protein binding is reduced by uremia.
Induction Agents and Sedatives
Although now rarely used, sodium thiopental serves as a good illustrative example of how reduced protein binding in CKD may affect the clinical use of an anesthetic agent. Burch and Stanski38 showed that the free fraction of an induction dose of thiopental is almost doubled in patients with renal failure. This accounts for the exaggerated clinical effects seen with thiopental in CKD patients and the substantial reduction in the necessary induction dose of this agent in uremic patients when compared with patients with normal renal function.
Ketamine is less extensively protein-bound than thiopental, and renal failure appears to have less influence on its free fraction. Redistribution and hepatic metabolism are largely responsible for termination of the anesthetic effects, with <3% of the drug excreted unchanged in the urine. Norketamine, the major metabolite, has one-third the pharmacologic activity of the parent drug and is further metabolized before it is excreted by the kidney.39
Etomidate, although only 75% protein-bound in normal patients, has a larger free fraction in patients with ESRD.40 The decrease in protein binding does not seem to alter the clinical effects of an etomidate anesthetic induction in patients with renal failure.
Propofol undergoes extensive, rapid hepatic biotransformation to inactive metabolites that are renally excreted. Its pharmacokinetics appear to be unchanged in patients with renal failure,41 and there are no reports of prolongation of its effects in ESRD.
The benzodiazepines, as a group, are extensively protein-bound. CKD increases the free fraction of benzodiazepines in the plasma, and this potentiates their clinical effect. Certain benzodiazepine metabolites are pharmacologically active and have the potential to accumulate with repeated administration of the parent drug to anephric patients. For example, 60% to 80% of midazolam is excreted as its (active) α-hydroxy metabolite,42 which accumulates during long-term infusions in patients with renal failure.42 AKI appears to slow the plasma clearance of midazolam, whereas repeated diazepam or lorazepam administration in CKD may carry a risk of active metabolite-induced sedation. Alprazolam is one of the few drugs related to anesthesia practice that has undergone pharmacodynamic studies in patients with CKD. Schmith et al.43 found that when decreased protein binding and increased free fraction of alprazolam are taken into account, patients with CKD are actually more sensitive to its sedative effects than healthy persons.
Dexmedetomidine is primarily metabolized in the liver. Volunteers with renal impairment receiving dexmedetomidine experienced a long-lasting sedative effect than subjects with normal kidney function. The most likely explanation is that less protein binding of dexmedetomidine occurs in subjects with renal dysfunction.44
Opioids
Single-dose studies of the pharmacokinetics of morphine in renal failure demonstrate no alteration in its disposition. However, chronic administration results in accumulation of its 6-glucuronide metabolite, which has potent analgesic and sedative effects.45 There is also a decrease in protein binding of morphine in ESRD, which mandates a reduction in its initial dose. Meperidine is remarkable for its neurotoxic, renally excreted metabolite (normeperidine) and is not recommended for use in patients with poor renal function. Hydromorphone is metabolized to hydromorphone-3-glucuronide, which is excreted by the kidneys. This active metabolite accumulates in patients with renal failure and may cause cognitive dysfunction and myoclonus.46 Codeine also has the potential for causing prolonged narcosis in patients with renal failure and cannot be recommended for long-term use.45
Fentanyl appears to be a better choice of opioid for use in ESRD because of its lack of active metabolites, unchanged free fraction, and short redistribution phase.47 Small-to-moderate doses, titrated to effect, are well tolerated by uremic patients.
Alfentanil has been shown to have reduced protein binding but no change in its elimination half-life or clearance in ESRD and is extensively metabolized to inactive compounds.48 Therefore, caution should be exercised in administering a loading dose, but the total dose and infusion dose should be similar to those for patients with normal renal function. The free fraction of sufentanil is unchanged in ESRD; however, its pharmacokinetics are variable, and it has been reported to cause prolonged narcosis.49
Remifentanil is rapidly metabolized by blood and tissue esterases to a weakly active (about 4,600 times less potent) μ-opioid agonist and renally excreted metabolite, remifentanil acid. Renal failure has no effect on the clearance of remifentanil, but elimination of the principal metabolite, remifentanil acid, is markedly reduced. However, the clinical implications of this metabolite are likely limited.50
Muscle Relaxants
Muscle relaxants are the most likely group of drugs used in anesthetic practice to produce prolonged effects in ESRD because of their dependence on renal excretion (Table 49-4). Only succinylcholine, atracurium, cis-atracurium, and mivacurium appear to have minimal renal excretion of the unchanged parent compound. Most nondepolarizing muscle relaxants must be either hepatically excreted or metabolized to inactive forms in order to terminate their activity. Some muscle relaxants have renally excreted, active metabolites that may contribute to their prolonged duration of action in patients with ESRD. Although the following discussion focuses on the pharmacology of individual muscle relaxants, coexisting acidosis and electrolyte disturbances, as well as drug therapy (e.g., aminoglycosides, diuretics, immunosuppressants, magnesium-containing antacids), may alter the pharmacodynamics of muscle relaxants in patients with renal failure.51
Succinylcholine has a long history of use in CKD that has been somewhat confused by conflicting reports of plasma cholinesterase activity in renal failure.52,53 Provided the serum potassium concentration is not dangerously elevated, its use can be justified as part of a rapid-sequence induction technique because its duration of action in ESRD is not significantly prolonged. Use of a continuous infusion of succinylcholine, however, is more problematic because the major metabolite, succinylmonocholine, is weakly active and excreted by the kidney. Concern about the increase in serum potassium levels after succinylcholine administration (0.5 mEq/L in normal subjects) implies that the serum potassium should be normalized to the extent possible in patients with renal failure, but clinical experience has shown that the acute small increase in potassium following administration of succinylcholine is generally well tolerated in patients with chronically elevated serum potassium levels. Use of the long-acting muscle relaxants doxacurium, pancuronium, and pipecuronium might also be questioned in patients with known renal insufficiency. In a single-dose study of doxacurium, Cook et al.54 demonstrated an increased elimination half-life, reduced plasma clearance, and prolonged duration of effect in patients with renal failure. Similar findings have been reported for the pharmacokinetics of pipecuronium.
Table 49-4. Nondepolarizing Muscle Relaxants in Renal Failure | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Intermediate-acting muscle relaxants (atracurium, cis-atracurium, vecuronium, and rocuronium) have a distinct advantage in ESRD because of their shorter duration. The risk of a clinically significant, prolonged block is much reduced. Atracurium and its derivative, cis-atracurium, undergo enzymatic ester hydrolysis and spontaneous nonenzymatic (Hoffman) degradation with minimal renal excretion of the parent compound. Their elimination half-life, clearance, and duration of action are not affected by renal failure,55 nor have they been reported to cause prolonged clinical effects in ESRD. These characteristics strongly support their use in patients with renal disease. One potential concern is that an atracurium metabolite, laudanosine, may cause seizures in experimental animals and may accumulate with repeated dosing or continuous infusion.56 However, this has not been realized in intensive care patients with renal failure receiving prolonged infusions of atracurium. Consistent with its greater potency and lower dosing requirements, cis-atracurium metabolism results in lower laudanosine blood levels than does atracurium in ESRD patients.
The pharmacokinetics of vecuronium were initially reported as unchanged in renal failure, but it was later demonstrated that its duration of action was prolonged as a result of reduced plasma clearance and increased elimination half-life.55 In addition, the active metabolite, 3-desmethylvecuronium, was shown to accumulate in anephric patients receiving a continuous vecuronium infusion who subsequently had prolonged neuromuscular blockade. An intubating dose would be expected to last approximately 50% longer in patients with ESRD.57
Rocuronium, a rapid-onset muscle relaxant, has a pharmacokinetic profile in normal subjects similar to that of vecuronium.58 Single-dose pharmacokinetic studies in patients with renal failure have reported conflicting results. Szenohradszky et al.59 reported that renal failure increased the volume of distribution and elimination half-life of rocuronium but had no effect on its clearance. Cooper et al.60 found that its clearance was reduced, and the duration of block was widely variable in patients with renal failure, although the mean duration of relaxation and spontaneous recovery was not statistically different from that in control subjects.
The short-acting muscle relaxant mivacurium is enzymatically eliminated by plasma pseudocholinesterase at a somewhat slower rate than succinylcholine. Low pseudocholinesterase activity correlates with slower recovery from a bolus dose of mivacurium in anephric patients.53 The maintenance infusion dose has been reported to be both lower61 and similar62 to that in normal control subjects.
The pharmacokinetics of the clinically available anticholinesterases are affected by renal failure.63 They have a prolonged duration of action in ESRD because of their heavy reliance on renal excretion. The anticholinergic agents atropine and glycopyrrolate, used in conjunction with the anticholinesterases, are similarly excreted by the kidney. Therefore, no dosage alteration of the anticholinesterases is required when antagonizing neuromuscular blockade in patients with reduced renal function.
Diuretic Drugs: Effects and Mechanisms
Fluid overload occurs when salt or water intake exceeds renal and extrarenal losses and is characterized by increased total body water and usually sodium. Fluid overload may be evenly distributed among the body compartments (e.g., congestive heart failure), or the interstitial space may be increased while the circulating blood volume may be normal or even decreased (e.g., posttraumatic or postoperative fluid shifts). Edema results when Starling forces favor passage of fluid into the interstitial space. A variety of chronic medical conditions (congestive heart failure, renal failure, or hepatic cirrhosis) can lead to fluid overload and edema that may even require surgery to be delayed for treatment to reduce operative risk. The first line of therapy for fluid overload that includes all body compartments involves restriction of salt and water ingestion; however, diuretic therapy is often indicated.
The Physiologic Basis of Diuretic Action
Diuretics are typically grouped according to their site and mechanism of action (Fig. 49-5). Under normal conditions, kidney function assures that <1% of the filtered Na+ load enters the urine (i.e., the fractional excretion of Na+ (FENa) is <1%). The Na+/K+-ATPase pump on the basolateral surface (blood side) of renal tubular cells is primarily responsible for active pumping of Na+ out of cells into blood in exchange for K+. This pump causes a net movement of positive charge out of the cell (2 K+ in, for every 3 Na+ out) creating an electrochemical gradient that also causes Na+ to enter the luminal (urine) side of the cell. Renal tubular cells in different portions of the nephron have different luminal “systems” to allow this Na+ influx. These systems are the sites of action where the different diuretics work.
Proximal Tubule Diuretics
In the proximal tubule, a specialized luminal transporter exchanges protons (H+) for sodium ions; the result is sodium reabsorption and acidification of the urine. The excreted H+ combines with bicarbonate (HCO3−) in the tubule to form carbonic acid: H+ + HCO3− → H2CO3. Carbonic acid converts to water (H2O) and carbon dioxide (CO2) in a reaction catalyzed by carbonic anhydrase: H2CO3 → H2O + CO2. The same enzyme, carbonic anhydrase, allows this reaction to occur in reverse within tubular cells, converting H2CO3 to HCO3− and H+, generating more H+ for countertransport with Na+, and releasing bicarbonate that passes into the circulation. Carbonic anhydrase inhibitors are drugs that inhibit this enzyme; the net effect of these agents is that sodium and bicarbonate, that would otherwise have been reabsorbed, remain in the urine and result in an alkaline diuresis.
Although patients may develop a metabolic acidosis when taking these agents, compensatory processes in the tubules accommodate the effects of carbonic anhydrase inhibitors so that their long-term use rarely causes this problem. However, these agents can be useful, for example, with contraction alkalosis from aggressive diuresis with loop diuretics (see later discussion); administration of these drugs can reduce PaCO2 and improve PaO2 for patients with little accompanying change in blood pH. Specific uses for carbonic anhydrase inhibitors include the treatment of mountain sickness, open-angle glaucoma, and to increase respiratory drive in patients with central sleep apnea.64,65
Osmotic Diuretics
Substances such as mannitol that are freely filtered at the glomerulus but poorly reabsorbed by the renal tubule will cause an osmotic diuresis. In the water-permeable segments of the proximal tubule and loop of Henle, fluid reabsorption occurs, and filtered mannitol is concentrated. Eventually oncotic pressure in the tubular fluid resists further fluid reabsorption. Mannitol also draws water from cells into the plasma and effectively increases RBF.
Mannitol has been widely used, especially for the prophylaxis of specific types of acute renal failures (ARFs) and in the treatment of increased intracranial pressure (see Chapter 36). In select patient populations, such as cadaveric kidney transplant recipients, it has been found to be effective.66 However, in a controlled trial of mannitol prophylaxis in patients with mild chronic renal failure, it was less effective than hydration alone for prevention of contrast-associated nephropathy.67 Although animal studies
showed initial promise, apart from AKI prophylaxis in kidney transplantation, there is no clear evidence that mannitol is effective either for the prevention or treatment of AKI.68 As mannitol shifts water between fluid compartments, there can be effects on plasma and intracellular electrolyte concentrations, including hyponatremia and hypochloremia and intracellular increases in K+ and H+. Patients with normal renal function quickly correct these changes, but patients with renal impairment may develop significant circulatory overload with hemodilution and pulmonary edema, hyperkalemic metabolic acidosis, central nervous system depression, and even severe hyponatremia requiring urgent hemodialysis.69
showed initial promise, apart from AKI prophylaxis in kidney transplantation, there is no clear evidence that mannitol is effective either for the prevention or treatment of AKI.68 As mannitol shifts water between fluid compartments, there can be effects on plasma and intracellular electrolyte concentrations, including hyponatremia and hypochloremia and intracellular increases in K+ and H+. Patients with normal renal function quickly correct these changes, but patients with renal impairment may develop significant circulatory overload with hemodilution and pulmonary edema, hyperkalemic metabolic acidosis, central nervous system depression, and even severe hyponatremia requiring urgent hemodialysis.69
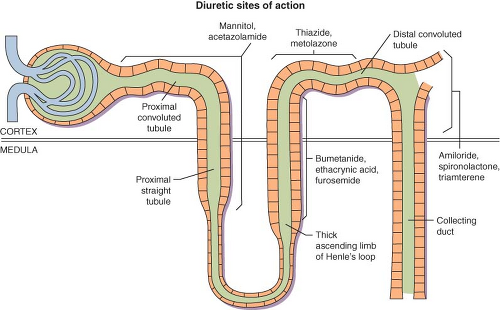 Figure 49.5. Site of action of commonly available diuretics. (From: Mende CW: Current issues in diuretic therapy. Hosp Pract. 1990;25(suppl 1):15, with permission.) |
Loop Diuretics
The electrochemical gradient established by the Na+/K+-ATPase in the loop of Henle drives the transport of one Na+, one K+, and two Cl− ions into the tubule cells from the tubular fluid. Because the thick ascending limb segment of the loop of Henle is water-impermeable, reabsorption of solute concentrates the interstitium and dilutes the tubular fluid. Loop diuretics, such as furosemide, bumetanide, and torsemide, directly inhibit the electroneutral transporter, preventing salt reabsorption from occurring. Because 25% of filtered NaCl is normally reabsorbed in the loop of Henle, loop diuretics cause a large salt load to pass to the distal convoluted tubule that is beyond the extra reserve of this tubular segment to reabsorb; consequently, large volumes of dilute urine ensue.
Loop diuretics are a first-line therapeutic modality for treatment of acute decompensated congestive heart failure. Although loop diuretics have no proven mortality benefit, they reduce left ventricular filling pressures and very effectively relieve the symptoms of congestion, pulmonary edema, extremity swelling, and hepatic congestion. Adverse effects of loop diuretics include hypokalemia, hyponatremia, and also AKI. Heart failure patients with atrial fibrillation may also be prescribed digitalis, which in combination with furosemide, can lead to hypokalemia-induced dysrhythmias. Loop diuretics, especially furosemide, may cause ototoxicity particularly in patients with renal insufficiency.70
Distal Convoluted Tubule Diuretics
Distal convoluted tubule diuretics, such as thiazides (e.g., hydrochlorothiazide) and metolazone, act in the early part of this segment to block the NaCl cotransport mechanism across apical plasma membranes. Because the distal tubule is relatively water-impermeable, net NaCl absorption causes urinary dilution. Clinically, distal convoluted tubule diuretics are used for the treatment of hypertension (often as sole therapy), volume overload disorders, and to relieve the symptoms of edema in pregnancy.
Adverse reactions associated with distal tubule diuretics include electrolyte disturbances and volume depletion. Hydrochlorothiazide specifically has been associated with a number of other side effects including pancreatitis, jaundice, diarrhea, and aplastic anemia.
Distal (Collecting Duct) Acting Diuretics
Unlike in the more proximal nephron segments, NaCl absorption in the collecting duct cells is not electroneutral. That is, a net electrical gradient is maintained both by the Na+/K+-ATPase Na+ ion channels and in the luminal membranes. As a result, the tubule lumen is negatively charged with respect to the blood. This normally causes K+ secretion into the tubular lumen through K+-specific ion channels. Distal K+-sparing diuretics (e.g., amiloride and triamterene) directly inhibit luminal Na+ entry, blocking this
mechanism, and resulting in a K+-“sparing” effect. In addition, H+ secretion is inhibited.
mechanism, and resulting in a K+-“sparing” effect. In addition, H+ secretion is inhibited.
A second class of distal-acting, potassium-sparing diuretics is the competitive aldosterone antagonists (e.g., spironolactone and eplerenone). Ordinarily, the mineralocorticoid hormone aldosterone is released by the body in response to angiotensin II or hyperkalemia. Aldosterone normally stimulates Na+ reabsorption and K+ excretion by the collecting duct. Inhibition of the aldosterone effect by these drugs causes a mild natriuresis and K+ retention. Distal K+-sparing agents are used primarily for K+-sparing diuresis (e.g., in patients with volume overload receiving digitalis or with hypokalemic alkalosis). In addition, these drugs are especially useful in treating disorders involving secondary hyperaldosteronism, such as cirrhosis with ascites. Spironolactone treatment has been shown to improve survival with volume overload and left ventricular dysfunction or heart failure.71 Hyperkalemia and hyperkalemic, hyperchloremic metabolic acidoses are significant complications of the injudicious use of spironolactone, triamterene, or amiloride.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


