Intravenous Anesthetics
Paul F. White
Matthew R. Eng
Key Points
Related Matter
Drug Concentration and Effect
Introduction
central nervous system (CNS) depression. This chapter focuses on the pharmacologic properties and clinical uses of the currently available IV anesthetics.
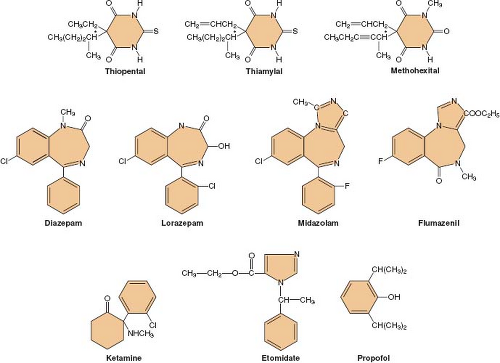 Figure 18.1. Chemical structures of currently available nonopioid intravenous anesthetics. |
Following its introduction into clinical practice, thiopental quickly became the gold standard of IV anesthetics against which all the newer IV drugs were compared. Many different hypnotic drugs are currently available for use during IV anesthesia (Fig. 18-1). Although propofol (currently the most widely used IV anesthetic) is both safe and effective, the “ideal” IV anesthetic is yet to be developed. The physical and pharmacologic properties that an ideal IV anesthetic would possess include the following:
Drug compatibility (water-solubility) and stability in solution.
Lack of pain on injection, veno-irritation, and local tissue damage following extravasation.
Low potential to release histamine or precipitate hypersensitivity reactions.
Rapid and smooth onset of hypnotic action without excitatory activity.
Rapid metabolism to pharmacologically inactive metabolites.
A steep dose-response relationship to enhance titratability and minimize tissue accumulation.
Lack of acute cardiovascular and respiratory depression.
Decreases in cerebral metabolism and intracranial pressure.
Rapid and smooth return of consciousness and cognitive skills with residual analgesia.
Absence of postoperative nausea and vomiting, amnesia, psychomimetic reactions, dizziness, headache, or prolonged sedation (“hangover” effects).
Historical Perspective
Despite thiopental’s proven clinical usefulness, safety, and widespread use over many decades of use, it has been supplanted by a variety of agents from different pharmacologic drug groups. The sedative–hypnotic drugs that have been more recently introduced into clinical practice (e.g., midazolam, ketamine, etomidate, propofol) have proven to be extremely valuable in specific clinical situations. These newer compounds combine many of the characteristics of the ideal IV anesthetic, but fail in aspects where the other drugs succeed. For some of these IV sedative–hypnotics, disadvantages have led to “restricted” indications (e.g., ketamine, etomidate). Although propofol is clearly the most popular of the sedative–hypnotic drug class, the optimal pharmacologic
properties are not equally important in every clinical situation. Therefore, the anesthesiologist must make the choice of the IV anesthetic drug that best fits the needs of the individual patient and the operative (or diagnostic) procedure.
properties are not equally important in every clinical situation. Therefore, the anesthesiologist must make the choice of the IV anesthetic drug that best fits the needs of the individual patient and the operative (or diagnostic) procedure.
General Pharmacology of Intravenous Anesthetics
Mechanism of Action
A widely accepted theory of anesthetic action is that both IV and inhalational anesthetics exert their primary sedative and hypnotic effects through an interaction with the inhibitory γ-aminobutyric acid (GABA) neurotransmitter system.3 GABA is the principal inhibitory neurotransmitter within the CNS. The GABA and adrenergic neurotransmitter systems counterbalance the action of excitatory neurotransmitters. The GABA type A (GABAA) receptor is a receptor complex consisting of up to five glycoprotein subunits. When the GABAA receptor is activated, transmembrane chloride conductance increases, resulting in hyperpolarization of the postsynaptic cell membrane and functional inhibition of the postsynaptic neuron. Sedative–hypnotic drugs interact with different components of the GABA-receptor complex (Fig. 18-2). However, the allosteric (structural) requirements for activation of the receptor are different for IV and volatile anesthetics.
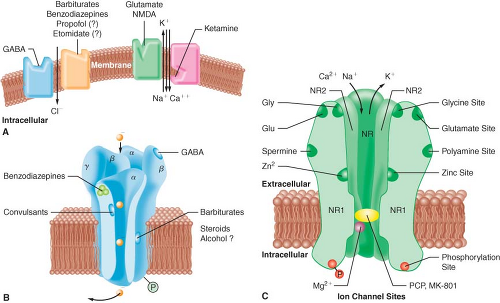 Figure 18.2. A: This model depicts the postsynaptic site of γ-aminobutyric acid (GABA) and glutamate within the central nervous system (CNS). GABA decreases the excitability of neurons by its action at the GABAA-receptor complex. When GABA occupies the binding site of this complex, it allows inward flux of chloride ion, resulting in hyperpolarizing of the cell and subsequent resistance of the neuron to stimulation by excitatory transmitters. Barbiturates, benzodiazepines, propofol, and etomidate decrease neuronal excitability by enhancing the effect of GABA at this complex, facilitating this inhibitory effect on the postsynaptic cell. Glutamate and its analog N-methyl-D-aspartate (NMDA) are excitatory amino acids. When glutamate occupies the binding site on the NMDA subtype of the glutamate receptor, the channel opens and allows Na+, K+, and Ca2+ to either enter or leave the cell. Flux of these ions leads to depolarization of the postsynaptic neuron and initiation of an action potential and activation of other pathways. Ketamine blocks this open channel and prevents further ion flux, thus inhibiting the excitatory response to glutamate. (Reprinted with permission from: Van Hemelrijck J, Gonzales JM, White PF. Use of intravenous sedative agents. In: Rogers MC, Tinker JH, Covino BG, Longnecker DE, eds. Principles and Practice of Anesthesiology. Mosby: St. Louis; 1992:1131.) B: Schematic model of the GABAA-receptor complex illustrating recognition sites for many of the substances that bind to the receptor. C: Model of the NMDA receptor showing sites for antagonist action. Ketamine binds to the site labeled PCP (phencyclidine). The pentameric structure of the receptor, composed of a combination of the subunits NR 1 and NR 2, is illustrated. (Altered with permission from: Leeson PD, Iversen LL. The glycine site on the NMDA receptor: Structure-activity relationships and therapeutic potential. J Med Chem. 1994;37:4053.) |
Benzodiazepines bind to specific receptor sites that are part of the GABAA-receptor complex. The binding of benzodiazepines to their receptor site increases the efficiency of the coupling between the GABA receptor and the chloride ion channel. The degree of modulation of the GABA-receptor function is limited, which explains the maximal “ceiling effect” produced by benzodiazepines with respect to CNS depression. The dose-dependent CNS depressant effect of benzodiazepines produce hypnosis, sedation, anxiolysis, amnesia, and anticonvulsant effects.1 These CNS effects are presumed to be associated with stimulation of
different receptor subtypes and/or concentration-dependent receptor occupancy.3 For example, it has been suggested that benzodiazepine receptor occupancy of 20% provides anxiolysis, while 30% to 50% receptor occupancy is associated with amnesia to sedation, and 60% receptor occupancy is required for hypnosis (or unconsciousness).1
different receptor subtypes and/or concentration-dependent receptor occupancy.3 For example, it has been suggested that benzodiazepine receptor occupancy of 20% provides anxiolysis, while 30% to 50% receptor occupancy is associated with amnesia to sedation, and 60% receptor occupancy is required for hypnosis (or unconsciousness).1
The interaction of barbiturates and propofol with specific membrane structures appears to decrease the rate of dissociation of GABA from its receptor, thereby increasing the duration of the GABA-activated opening of the chloride ion channel (Fig. 18-2). Barbiturates can also mimic the action of GABA by directly activating the chloride channels. The proposed mechanism of action of thiopental relates to its ability to function as a competitive inhibitor at the nicotinic acetylcholine receptors in the CNS.4 Etomidate augments GABA-gated chloride currents (i.e., indirect modulation) and at higher concentrations evokes chloride currents in the absence of GABA (i.e., direct activation). Although the mechanism of action of propofol is similar to that of the barbiturates (i.e., enhancing the activity of the GABA-activated chloride channel), it also possesses ion channel-blocking effects in cerebral cortex tissue and nicotinic acetylcholine receptors, as well as an inhibitory effect on lysophosphatidate signaling in lipid mediator receptors.5
Ketamine produces a functional dissociation between the thalamocortical and limbic systems, a state that has been termed dissociative anesthesia. Ketamine depresses neuronal function in the cerebral cortex and thalamus, while simultaneously activating the limbic system. The effect of ketamine on the medial medullary reticular formation may be involved in the affective component of its nociceptive activity. The CNS effects of ketamine appear to be primarily related to its antagonistic activity at the N-methyl-D-aspartate (NMDA) receptor (Fig. 18-2). Unlike the other IV anesthetics, ketamine does not interact with GABA receptors; however, it binds to non-NMDA glutamate receptors and nicotinic, muscarinic, monoaminergic, and even kappa-opioid receptors. In addition, it also inhibits neuronal sodium channels (producing a modest local anesthetic action) and calcium channels (causing cerebral vasodilatation).
Table 18-1. Pharmacokinetic Values for the Currently Available Intravenous Sedative–Hypnotic Drugs | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Pharmacokinetics and Metabolism
An understanding of basic pharmacokinetic principles is integral to the understanding of the pharmacologic actions and interactions of IV anesthetic and adjunctive drugs, and will allow the anesthesiologist to develop more optimal dosing strategies when using IV techniques (see Chapter 7 Basic Principles of Clinical Pharmacology). Although lipid solubility facilitates diffusion of IV anesthetics across cellular membranes, including the blood–brain barrier, only the nonionized form of the compound is able to readily cross neuronal membranes. The ratio of the unionized-to-ionized fraction depends on the pKa of the drug and the pH of the body fluids.
The rapid onset of the CNS effect of most IV anesthetics can be explained by their high lipid solubility and the relatively high proportion of the cardiac output (20%) perfusing the brain. However, a variable degree of hysteresis exists between the blood concentration of the hypnotic drug and its onset of action on the CNS. The hysteresis is related in part to diffusion of these drugs into brain tissue and nonspecific CNS receptor binding. However, the number of CNS binding sites is usually saturable and only a small fraction of the available binding sites needs to be occupied to produce clinical effects. Although the total amount of drug in the blood is available for diffusion, the diffusion rate will be more limited for IV anesthetics with a high degree of plasma protein binding (90%) because only the “free” unbound drug can diffuse across membranes and exert central effects. When several drugs compete for the same binding sites, or when the protein concentration in the blood is decreased by preexisting disease (e.g., hepatic failure, malnutrition), a higher fraction of the unbound drug will be available to exert an effect on the CNS. Since only unbound drug is available for uptake and metabolism in the liver, highly protein-bound drugs may have a lower rate of hepatic metabolism as a result of their decreased hepatic extraction ratio (i.e., the fraction of the hepatic blood flow that is cleared of the drug).
The pharmacokinetics of IV hypnotics are characterized by rapid distribution and subsequent redistribution into several
hypothetical compartments (determined by their effect on blood flow to various tissues), followed by elimination (Table 18-1). The initial pharmacologic effects are related to the activity of the drug in the central compartment. The primary mechanism for terminating the central effects of IV anesthetics administered for induction of anesthesia is redistribution from the central highly perfused compartment (e.g., brain) to the larger, but less well perfused “peripheral” compartments (e.g., muscle, fat). Even for drugs with a high hepatic extraction ratio (e.g., propofol), elimination does not always play a major role in terminating the drug’s sedative–hypnotic effects because elimination of the drug can occur only from the central compartment. The rate of elimination from the central compartment, the amount of drug present in the peripheral compartments, and the rate of redistribution from the peripheral compartments “back” into the central compartment determine the time necessary to eliminate the drug from the body and can directly influence intermediate and late recovery times.
hypothetical compartments (determined by their effect on blood flow to various tissues), followed by elimination (Table 18-1). The initial pharmacologic effects are related to the activity of the drug in the central compartment. The primary mechanism for terminating the central effects of IV anesthetics administered for induction of anesthesia is redistribution from the central highly perfused compartment (e.g., brain) to the larger, but less well perfused “peripheral” compartments (e.g., muscle, fat). Even for drugs with a high hepatic extraction ratio (e.g., propofol), elimination does not always play a major role in terminating the drug’s sedative–hypnotic effects because elimination of the drug can occur only from the central compartment. The rate of elimination from the central compartment, the amount of drug present in the peripheral compartments, and the rate of redistribution from the peripheral compartments “back” into the central compartment determine the time necessary to eliminate the drug from the body and can directly influence intermediate and late recovery times.
Most IV anesthetic agents are eliminated via hepatic metabolism followed by renal excretion of more water-soluble metabolites. Some metabolites have pharmacologic activity and can produce prolonged drug effects (e.g., oxazepam, desmethyldiazepam, norketamine). Moreover, there is considerable interpatient variability in the clearance rates for commonly used IV anesthetic drugs. The elimination clearance is the distribution volume cleared of drug over time and is a measure of the efficacy of the elimination process. The slow elimination of some anesthetics is partly due to their high degree of protein binding that reduces their hepatic extraction ratio. Other drugs may have a high hepatic extraction ratio and elimination clearance despite extensive plasma protein binding (e.g., propofol), indicating that protein binding is not always a rate-limiting factor.
When a drug infusion is administered without a loading dose, at least three times the t1/2β value may be required to achieve a true “steady state” plasma concentration. The steady state concentration obtained during an anesthetic infusion depends on the rate of drug administration and its clearance rate. When an infusion is discontinued, the rate at which the plasma concentration decreases largely depends on the clearance rate (as reflected by the terminal t1/2β value). For drugs with shorter elimination half-lives, plasma concentration will decrease at a rate that allows for a more rapid recovery (e.g., propofol). Drugs with longer t1/2β values (e.g., thiopental and diazepam) are usually only administered by continuous IV infusion when the medical condition requires long-term treatment (e.g., elevated intracranial pressure [ICP] as a result of brain injury or prolonged sedation in the intensive care unit [ICU] because of respiratory failure).
Careful titration of an anesthetic drug to achieve the desired clinical effect is necessary to avoid drug accumulation and the resultant prolonged CNS effects after the infusion has been discontinued. Although the value of the t1/2β indicates how fast a drug is eliminated from the body, a more useful indicator of the acceptability of a hypnotic infusion for maintenance of anesthesia or sedation is the so-called context-sensitive half-time, a value derived from computer simulations of drug infusions.11 The context-sensitive half-time is defined as the time necessary for the effect-compartment (i.e., effect site) concentration to decrease by 50% in relation to the duration of the infusion. The context-sensitive half-time becomes particularly important in determining recovery after prolonged infusions of sedative–hypnotic drugs. Drugs (e.g., propofol) may have a relatively short context-sensitive half-time despite the fact that a large amount of drug remains present in the “deep” (less well-perfused) tissue compartment. The slow return of the anesthetic from the deep compartment contributes little to the concentration of drug in the central compartment from which it is rapidly cleared. Therefore, the concentration in the central compartment rapidly declines below the hypnotic threshold after discontinuation of the infusion, contributing to short emergence times despite the fact that a substantial quantity of anesthetic drug may remain in the body.
Marked interpatient variability exists in the pharmacokinetics of IV sedative–hypnotic drugs. Factors that can influence anesthetic drug disposition include the degree of protein binding, the efficiency of hepatic and renal elimination processes, physiologic changes with aging, preexisting disease states, the operative site, body temperature, and drug interactions (e.g., coadministration of volatile anesthetics). For example, increased age, lean body (muscle) mass, and total body water decrease result in an increase in the steady state volume of distribution of most IV anesthetics. The increased distribution volume and decreased hepatic clearance leads to a prolongation of their t1/2β values. Moreover, a decrease of the volume of the central compartment may result in higher initial drug concentrations and can at least partially explain the decreased induction requirement in the elderly. Additionally, the slower redistribution from the vessel-rich tissues to intermediate compartments (e.g., muscles) also contributes to the age-related decrease in the induction dose requirements.11 Although prolongation of the elimination half-time in the elderly does not provide an explanation for the decreased induction dose requirement, it is responsible for producing higher steady state plasma concentrations of IV anesthetics at any given infusion rate when administered for maintenance of general anesthesia or sedation, contributing to a slower recovery from the sub-hypnotic (residual) CNS depressant effects.
The hepatic clearance of IV anesthetics with a high (e.g., etomidate, propofol, ketamine) or intermediate (e.g., methohexital, midazolam) extraction ratio largely depends on hepatic blood flow, with most of the drug being removed from the blood as it flows through the liver (so-called perfusion-limited clearance). The elimination rate of drugs with low hepatic extraction ratios (e.g., thiopental, diazepam, lorazepam) depends on the enzymatic activity of the liver and is less dependent of hepatic blood flow (so-called capacity-limited clearance). Hepatic blood flow decreases during upper abdominal and laparoscopic surgery and, as a result, higher blood levels of drugs with perfusion-limited clearance are achieved at any given infusion rate. With aging, a decreased cardiac output and a redistribution of blood flow can partly explain the lower clearance rate for drugs with perfusion-limited clearance. Although concomitant administration of volatile anesthetics (which are known to decrease liver blood flow) has little influence on the elimination of thiopental, they can decrease the clearance of etomidate, ketamine, methohexital, and propofol. Other factors that decrease hepatic blood flow include hypocapnia, congestive heart failure, intravascular volume depletion, acute alcohol intoxication, circulatory collapse, increase intra-abdominal pressure, β-adrenergic blockade, and norepinephrine administration.
Hepatic disease can influence the pharmacokinetics of drugs by: (1) altering the plasma protein content and changing the degree of protein binding, (2) decreasing hepatic blood flow and producing intrahepatic shunting, and (3) depressing the metabolic enzymatic activity of the liver. Therefore, the influence of hepatic disease on pharmacokinetics and dynamics of IV anesthetics is difficult to predict. Renal disease can also alter the concentration of plasma and tissue proteins, as well as the degree of protein binding, thereby producing changes in free drug concentrations. Because IV anesthetic agents are primarily metabolized by the liver, renal insufficiency has little influence on their rate of metabolic inactivation or elimination of the primary compound.
Pharmacodynamic Effects
The principal pharmacologic effect of IV anesthetics is to produce progressively increasing sedation and ultimately hypnosis as a result of dose-dependent CNS depression. However, all sedative–hypnotics also directly or indirectly affect other major organ systems. The relationship between the dose of a sedative–hypnotic and its CNS effects can be defined by dose-response curves. Although most IV anesthetics are characterized by steep dose-response curves, the slopes of the curves are not always parallel (Fig. 18-3). However, the characteristics of a dose-response curve can only be interpreted in relation to the specific response for which it was constructed.
When steady state plasma concentrations are achieved, one can assume that the plasma concentration is in quasi-equilibrium with the effect-site concentration. Under these circumstances, it is possible to describe the relationship between drug and effect using a concentration-effect curve (Fig. 18-4). Because of the pharmacodynamic variability that exists among individuals, the plasma drug concentration necessary to obtain a particular effect is often described in terms of an effective concentration range, the so-called therapeutic window. Efficacy of an IV anesthetic relates to the maximum effect that can be achieved with respect to some measure of CNS function. Depending on the drug effect under consideration, the efficacy of sedative–hypnotics may appear to be <100%. Compared to thiopental and propofol, it is extremely difficult to produce an isoelectric [silent] EEG pattern with a benzodiazepine. Potency, on the other hand, relates to the quantity of drug necessary to obtain the maximum CNS effect. The relative potency of sedative–hypnotics also varies depending on the end point chosen. In the presence of an antagonist drug (e.g., flumazenil), the maximal response that can be obtained with a benzodiazepine agonist is further reduced because of competition for the same CNS receptor binding sites.
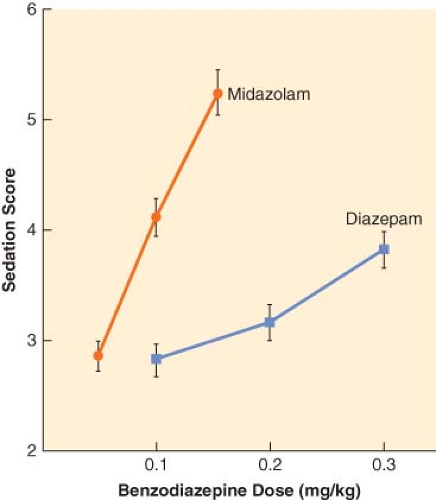 Figure 18.3. Dose-response relationships for sedation with midazolam (•) and diazepam (▪). The level of sedation (2 = awake and alert to 6 = asleep and unarousable) was assessed 5 minutes after bolus doses of midazolam (0.05, 0.1, or 0.15 mg/kg) or diazepam (0.1, 0.2, or 0.3 mg/kg). Values represent mean values ± SEM. (Reprinted with permission from: White PF, Vasconez LO, Mathes SA, et al. Comparison of midazolam and diazepam for sedation during plastic surgery. J Plast Reconstruct Surg. 1988;81:703.) |
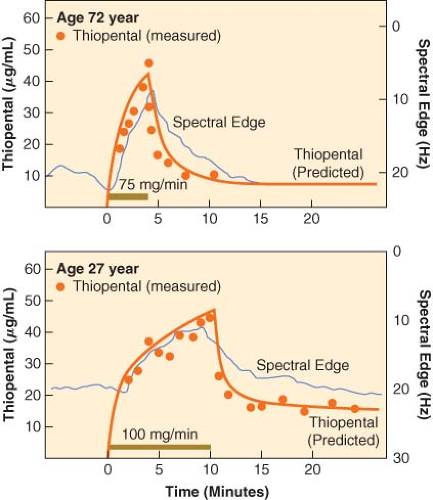 Figure 18.4. The concentration of thiopental versus time and spectral edge in an elderly patient (top) and in a younger patient (bottom). Solid horizontal bars represent the length of thiopental infusion. Filled circles represent the measured thiopental concentration (linear scale), and the solid line next to them represents the fitted data from the pharmacokinetic model. The axis for spectral edge has been inverted for visual clarity. (Reprinted with permission from: Homer TD, Stanski DR. The effect of increasing age on thiopental disposition and anesthetic requirement. Anesthesiology. 1985;62:714.) |
The influence of sedative–hypnotics on cerebral metabolism, cerebral hemodynamics, and ICP is of particular importance during neuroanesthesia. In patients with reduced cerebral compliance, a small increase in cerebral blood volume can cause a life-threatening increase in ICP. Most sedative–hypnotic drugs cause a proportional reduction in cerebral metabolism
(CMRO2) and cerebral blood flow (CBF), resulting in a decrease in ICP. Although a decrease in CMRO2 probably provides only a modest degree of protection against CNS ischemia or hypoxia, some hypnotics appear to possess cerebroprotective potential (e.g., thiopental, propofol) (see also Chapter 36 Anesthesia for Neurosurgery). Explanations for the alleged neuroprotective effects of these compounds include a biochemical role as free radical scavengers and membrane stabilizers (barbiturates and propofol) or NMDA-receptor antagonists (ketamine). With the exception of ketamine, all sedative–hypnotics also lower intraocular pressure. The changes in intraocular pressure generally reflect the effects of the IV agent on systemic arterial pressure and intracranial hemodynamics. However, none of the available sedative–hypnotic drugs protect against the transient increase in intraocular pressure that occurs with laryngoscopy and tracheal intubation.
(CMRO2) and cerebral blood flow (CBF), resulting in a decrease in ICP. Although a decrease in CMRO2 probably provides only a modest degree of protection against CNS ischemia or hypoxia, some hypnotics appear to possess cerebroprotective potential (e.g., thiopental, propofol) (see also Chapter 36 Anesthesia for Neurosurgery). Explanations for the alleged neuroprotective effects of these compounds include a biochemical role as free radical scavengers and membrane stabilizers (barbiturates and propofol) or NMDA-receptor antagonists (ketamine). With the exception of ketamine, all sedative–hypnotics also lower intraocular pressure. The changes in intraocular pressure generally reflect the effects of the IV agent on systemic arterial pressure and intracranial hemodynamics. However, none of the available sedative–hypnotic drugs protect against the transient increase in intraocular pressure that occurs with laryngoscopy and tracheal intubation.
Most IV hypnotics have similar EEG effects. Activation of high-frequency EEG activity (15 to 30 Hz) is characteristic of low concentrations (so-called sedative doses) of IV anesthetics. At higher concentrations, an increase in the relative contribution of the lower frequency higher amplitude waves is observed. At high concentrations, a burst-suppressive pattern develops with an increase in the isoelectric periods. Most sedative–hypnotic drugs have been reported to cause occasional EEG seizure-like myoclonic activity. Interestingly, these same drugs also possess anticonvulsant properties.12,13 When considering possible epileptogenic properties of CNS-depressant drugs, it is important to differentiate between true epileptogenic activity (e.g., methohexital) and myoclonic-like phenomena (e.g., etomidate, ketamine, propofol). Myoclonic activity is generally considered to be the result of an imbalance between excitatory and inhibitory subcortical centers, produced by an unequal degree of suppression of these brain centers by low concentrations of hypnotic drugs. Epileptic activity refers to a sudden alteration in CNS seizure-like activity resulting from a high-voltage electrical discharge at either cortical or subcortical sites, with subsequent spreading to the thalamic and brainstem centers. As a result of its vasoconstrictive effects on the cerebral vasculature, propofol may be useful for treatment of intractable migraine headaches.14
Although some induction drugs can increase airway sensitivity, coughing and airway irritation (e.g., bronchospasm) are usually a result of manipulation of the airway during “light” (inadequate) levels of IV anesthesia rather than to a direct drug effect. With the exception of ketamine (and to a lesser extent, etomidate), IV anesthetics produce dose-dependent respiratory depression, which is enhanced in patients with chronic obstructive pulmonary disease. The respiratory depression is characterized by a decrease in tidal volume and minute ventilation, as well as a transient rightward shift in the CO2 response curve. Following the rapid injection of a large bolus dose of an IV anesthetic, transient apnea lasting 30 to 90 seconds is usually produced. Ketamine causes minimal respiratory depression when administered in the usual induction doses, while etomidate is associated with less respiratory depressant effects than the barbiturate compounds or propofol. The α2-agonist dexmedetomidine has minimal depressant effects on respiratory function.15 The sympatholytic effects of dexmedetomidine when administered for premedication may increase the incidence of intraoperative hypotension and bradycardia.16
Many different factors contribute to the hemodynamic changes associated with IV induction of anesthesia, including the patient’s preexisting cardiovascular and fluid status, resting sympathetic nervous system tone, chronic cardiovascular drugs, preanesthetic medication, the speed of drug injection, and the onset of unconsciousness. In addition, cardiovascular changes can be attributed to the direct pharmacologic actions of anesthetic and analgesic drugs on the heart and peripheral vasculature. IV anesthetics can depress the CNS and peripheral nervous system responses, blunt the compensatory baroreceptor reflex mechanisms, produce direct myocardial depression, and lower peripheral vascular resistance (and/or dilate venous capacitance vessels), thereby decreasing venous return. Profound hemodynamic effects occur at induction of anesthesia in the presence of hypovolemia because a higher than expected drug concentration is achieved in the central compartment. Not surprisingly, the acute cardiocirculatory depressant effects of all IV anesthetics are accentuated in the elderly, as well as in the presence of preexisting cardiovascular disease (e.g., coronary artery disease, chronic hypertension).
Hypersensitivity (Allergic) Reactions
Allergic or hypersensitivity-type reactions to IV anesthetics are rare but can be severe and even life-threatening. IV drug administration bypasses the normal “protective barriers” against entrance of foreign molecules into the body. With the exception of etomidate, all IV induction agents have been alleged to cause some histamine release. However, the incidence of severe anaphylactic reactions is extremely low with the currently available IV induction agents. The high frequency of allergic reactions to the Cremophor EL–containing formulations led to the early withdrawal of IV anesthetics containing this solubilizing agent (e.g., propofol EL, propanidid, Alphadione [Althesin]). The possible mechanisms for immunologic reactions include: (1) direct action on mast cells, (2) classic complement activation after previous exposure and antibody formation, (3) complement activation through the alternative pathway without previous antigen exposure, (4) antigen-antibody reactions, and (5) the “mixed type” of anaphylactoid reactions.
Severe anaphylactic reactions to IV anesthetics are extremely uncommon; however, profound hypotension attributed to nonimmunologically mediated histamine release has been reported with thiopental use. Although anaphylactic reactions to etomidate have been reported, it does not appear to release histamine, and is considered to be the most “immunologically safe” IV anesthetic. Propofol does not normally trigger histamine release, but life-threatening anaphylactoid reactions have been reported in patients with a previous history of multiple-drug allergies. Barbiturates can also precipitate episodes of acute intermittent porphyria and their use is contraindicated in predisposed patients. Although benzodiazepines, ketamine, and etomidate are reported to be safe in humans, these drugs have been shown to be porphyrogenic in animal models. The most common causes of profound hypotension following IV induction of anesthesia are unexpected drug interactions and/or unrecognized hypovolemia.
Comparative Physicochemical and Clinical Pharmacologic Properties of Intravenous Agents
Barbiturates
The most commonly used barbiturates are thiopental (5-ethyl-5-[1-methylbutyl]-2-thiobarbituric acid), methohexital (1-methyl-5-allyl-5-[1-methyl-2-pentanyl] barbituric acid), and thiamylal (5-allyl-5-[1-methylbutyl]-2-thiobarbituric acid). Thiopental (Pentothal) and thiamylal (Surital) are thiobarbiturates, while methohexital (Brevital) is an oxybarbiturate. Thiamylal is slightly more potent than thiopental but has a similar pharmacologic profile. Although the l-isomers of thiopental and thiamylal are twice as potent as the d-isomers, both hypnotics are commercially available as racemic mixtures. Because methohexital has two asymmetric centers, it has four stereoisomers. The β-l-isomer is 4 to 5 times more potent than the α-l-isomer, but it produces excessive motor responses. Therefore, methohexital is marketed as the racemic mixture of the two α-isomers.
All three barbiturates are available as sodium salts and must be dissolved in isotonic sodium chloride (0.9%) or water to prepare solutions of 2.5% thiopental, 1% to 2% methohexital, and 2% thiamylal. If refrigerated, solutions of the thiobarbiturates are stable for up to 2 weeks. Solutions of methohexital are stable for up to 6 weeks. When barbiturates are added to Ringer lactate or an acidic solution containing other water-soluble drugs, precipitation will occur and can occlude the IV catheter. Although the typical solution of thiopental (2.5%) is highly alkaline (pH 9) and can be irritating to the tissues if injected extravenously, it does not cause pain on injection and venoirritation is rare. In contrast, a 1% methohexital solution frequently causes discomfort when injected into small veins. Intra-arterial injection of thiobarbiturates is a serious complication as crystals can form in the arterioles and capillaries, causing intense vasoconstriction, thrombosis, and even tissue necrosis. Accidental intra-arterial injections should be treated promptly with intra-arterial administration of papaverine and lidocaine (or procaine), as well as a regional anesthesia-induced sympathectomy (stellate ganglion block, brachial plexus block) and heparinization.
Thiopental is metabolized in the liver to hydroxythiopental and the carboxylic acid derivative, which are more water soluble and have little CNS activity. When high doses of thiopental are administered, a desulfuration reaction can occur with the production of pentobarbital, which has long-lasting CNS-depressant activity. The low elimination clearance of thiopental (3.4 mL/kg/min) contributes to a long elimination half-life (t1/2β of 11 hours). Preexisting hepatic and renal disease result in decreased plasma protein binding, thereby increasing the free fraction of thiopental and enhancing its CNS and cardiovascular-depressant properties. During prolonged continuous administration of thiopental, the concentration in the tissues approaches the concentration in the central compartment, with termination of its CNS effects becoming solely dependent on elimination by nonlinear hepatic metabolism. Methohexital is metabolized in the liver to inactive hydroxyderivatives. The clearance of methohexital (11 mL/kg/min) is higher and more dependent on hepatic blood flow than thiopental, resulting in a shorter elimination half-life (t1/2β 4 hours).
The usual induction dose of thiopental is 3 to 5 mg/kg in adults, 5 to 6 mg/kg in children, and 6 to 8 mg/kg in infants. Because methohexital is approximately 2.7 times more potent than thiopental, a dose of 1.5 mg/kg is equivalent to 4 mg/kg of thiopental in adults. The dose of barbiturates necessary to induce anesthesia is reduced in premedicated patients, patients in early pregnancy (7 to 13 weeks’ gestation), and those of more advanced American Society of Anesthesiologists physical status (III or IV). Geriatric patients require a 30% to 40% reduction in the usual adult dose because of a decrease of the volume of the central compartment and slowed redistribution of thiopental from the vessel-rich tissues to lean muscle.18 When the calculation of the induction dose is based on the lean body mass rather than total body weight, dosage adjustments for age, sex, or obesity are not necessary. Thiopental infusion is seldom used to maintain anesthesia because of the long context-sensitive half-time and prolonged recovery period. Plasma thiopental levels necessary to maintain a hypnotic state range between 10 and 20 mg/mL. A typical infusion rate necessary to treat intracranial hypertension or intractable convulsions is 2 to 4 mg/kg/hr. The plasma concentration of methohexital needed to maintain hypnosis during anesthesia ranges between 3 and 5 mg/mL and can be achieved with an infusion rate of methohexital 50 to 120 μg/kg/min.
Barbiturates produce a proportional decrease in CMRO2 and CBF, thereby lowering ICP. The maximal decrease in CMRO2 (55%) occurs when the EEG becomes isoelectric (burst-suppressive pattern). An isoelectric EEG can be maintained with a thiopental infusion rate of 4 to 6 mg/kg/hr (resulting in plasma concentrations of 30 to 50 μg/mL). Because the decrease in systemic arterial pressure is usually less than the reduction in ICP, thiopental should improve cerebral perfusion and compliance. Therefore, thiopental is widely used to improve brain relaxation during neurosurgery and to improve cerebral perfusion pressure (CPP) after acute brain injury. Although barbiturate therapy is widely used to control ICP after brain injury, the results of outcome studies are no better than with other aggressive forms of cerebral antihypertensive therapy.
It has been suggested that barbiturates also possess “neuroprotective” properties secondary to their ability to decrease oxygen demand. Alternative explanations have been suggested, including a reverse steal (“Robin Hood effect”) on CBF, free-radical scavenging, stabilization of liposomal membranes, as well as excitatory amino acid receptor blockade. On the basis of the evidence from experimental studies and a large randomized prospective multi-institutional study,19 experts have concluded that barbiturates have no place in the therapy following resuscitation of a cardiac arrest patient. In contrast, barbiturates are frequently used for cerebroprotection during incomplete brain ischemia (e.g., carotid endarterectomy, temporary occlusion of cerebral arteries, profound hypotension, and cardiopulmonary bypass). By improving the brain’s tolerance of incomplete ischemia in patients undergoing open heart surgery with cardiopulmonary bypass, barbiturates were alleged to decrease the incidence of postbypass neuropsychiatric disorders.20 However, during valvular open heart cardiac surgery, a protective effect of barbiturate loading could not be demonstrated.21 Given the lack of demonstrable neuroprotective effect, use of barbiturates during cardiac surgery is not recommended. Use of moderate degrees of hypothermia (33–34°C) might provide superior neuroprotection to the barbiturates without prolonging recovery.
EEG seizure discharges in patients with temporal lobe epilepsy. It is also the IV anesthetic of choice for electroconvulsive therapy22 because, compared to thiopental and propofol, methohexital produces less depression of EEG activity. Since the frequency of epileptiform EEG activity during induction of anesthesia with methohexital is significantly less than that which occurs during normal periods of sleep in epileptic patients, this suggests that higher doses of methohexital produces anticonvulsant activity. Methohexital also causes myoclonic-like muscle tremors and other signs of excitatory activity (e.g., hiccoughing).
Barbiturates cause dose-dependent respiratory depression.23 However, bronchospasm or laryngospasm following induction with thiopental is usually the result of airway manipulation in “lightly” anesthetized patients. Laryngeal reflexes appear to be more active after induction with thiopental than with propofol. The cardiovascular effects of thiopental and methohexital include decreases in cardiac output, systemic arterial pressure, and peripheral vascular resistance. The depressant effects of thiopental on cardiac output are primarily a result of a decrease in venous return caused by peripheral pooling, as well as a result of a direct myocardial depressant effect, which assumes increasing importance in the presence of hypovolemia and myocardial disease.24 Use of appropriate doses can minimize the cardiodepressant effects of thiopental, even in infants.25 An equipotent dose of methohexital produces even less hypotension than thiopental because of a greater tachycardic response to the blood pressure-lowering effects of the drug. If the blood pressure remains stable, the myocardial oxygen demand/supply ratio remains normal despite the increase in heart rate because of a concurrent decrease in coronary vascular resistance.
Propofol
Propofol (2,6-disopropylphenol), an alkylphenol compound, is virtually insoluble in aqueous solution. The initial Cremophor EL formulation of propofol was withdrawn from clinical testing because of the high incidence of anaphylactic reactions. Subsequently, propofol (10 mg/mL) was reintroduced as an egg lecithin emulsion formulation (Diprivan), consisting of 10% soybean oil, 2.25% glycerol, and 1.2% egg phosphatide. Microbial contamination of the original Diprivan formulation lead the manufacturer to add ethylenediaminetetraacetic acid (EDTA) to the propofol solution. With this formulation, injection pain occurs in 32% to 67% of patients when the drug is injected into small hand veins. This side effect can be minimized by injecting propofol into larger veins and by prior administration of either lidocaine or a potent opioid analgesic (e.g., fentanyl or remifentanil). A wide variety of drugs have been alleged to reduce pain on injection of propofol [e.g., metoprolol,26 granisetron,27 dolasetron,28 and even thiopental29]. Diluting the formulation with additional solvent (Intralipid) or changing the lipid carrier (Lipofundin) also reduced propofol-induced injection pain, probably because of a decrease in the concentration of free propofol in the aqueous phase of the emulsion. A new propofol formulation with sodium metabisulphite (instead of EDTA) as an antimicrobial has been shown to be associated with less severe pain on injection.30 Although the presence of the metabisulphite has raised concerns regarding its use in sulphite-allergic patients, this concern does not appear to be a clinically important problem. Of interest, a 2% formulation is available for long-term sedation to decrease the fluid (and lipid) volume.
A lower-lipid formulation of propofol (Ampofol) was introduced into clinical practice for both general anesthesia31 and sedation.32 The increased “free” fraction of propofol leads to increased pain when it is injected into small veins. Therefore, it is important to add lidocaine to the Ampofol formulation to minimize the pain on injection. A water-soluble prodrug of propofol, fospropofol (aka Aquavan or Lusedra), was approved by the FDA for use during local and regional anesthesia (e.g., conscious sedation and/or monitored anesthesia care [MAC]). This prodrug is rapidly hydrolyzed by plasma alkaline phosphatases in the circulation to release free propofol.33 Therefore, it does not produce the typical pain on injection associated with propofol. However, it has a slower onset than propofol34 and is frequently associated with a transient severe burning sensation in the perineal region immediately following IV injection. A microemulsion formulation of propofol (Aquafol) has been shown to be as effective and safe as the classical propofol formulation with similar pharmokinetics, but is associated with more severe and frequent injection pain.35 A new lipid-free preparation of propofol has been developed containing the drug, sulfobutylether β-cyclodextrin and water.36 Unfortunately, propofol in cyclodextrin (Captisol-enabled propofol) also produced significantly higher injection pain scores than the standard formulation.
Propofol’s pharmacokinetics has been studied using single-bolus dosing and continuous infusions.37 In studies using a two-compartment kinetic model, the initial distribution half-life is 2 to 4 minutes and the elimination half-life is 1 to 3 hours. Using a three-compartment model, the initial and slow distribution half-life values are 1 to 8 minutes and 30 to 70 minutes, respectively. The elimination half-life depends largely on the sampling time after discontinuing the administration of propofol and ranges from 2 to 24 hours. This long elimination half-life is indicative of the existence of a poorly perfused compartment from which propofol slowly diffuses back into the central compartment. Propofol is rapidly cleared from the central compartment by hepatic metabolism and the context-sensitive half-life for propofol infusions up to 8 hours is <40 minutes. Propofol is rapidly and extensively metabolized to inactive, water-soluble sulphate and glucuronic acid metabolites, which are eliminated by the kidneys. The clearance rate of propofol (20 to 30 mL/kg/min) exceeds hepatic blood flow, suggesting that an extrahepatic route of elimination (lungs) also contributes to its clearance. Nevertheless, changes in liver blood flow would be expected to produce marked alterations in the clearance rate of propofol. Surprisingly, few changes in propofol’s pharmacokinetics have been reported in the presence of hepatic or renal disease.
The induction dose of propofol in healthy adults is 1.5 to 2.5 mg/kg, with blood levels of 2 to 6 μg/mL producing unconsciousness depending on the concomitant medications (e.g., opioid analgesics), the patient’s age and physical status, and the extent of the surgical stimulation.38 In the morbidly obese patient, the propofol induction and maintenance dosages should be calculated on the basis of patient’s lean body weight.39 In one of the first reports describing the use of propofol for induction and maintenance of anesthesia with nitrous oxide, an average infusion rate of 120 μg/kg/min was used.40 The recommended maintenance infusion rate of propofol varies between 100 and 200 μg/kg/min for hypnosis and between 25 and 75 μg/kg/min for sedation. Awakening typically occurs at plasma propofol concentrations of 1 to 1.5 μg/mL.41 Because a 50% decrease in the plasma propofol concentration is usually required for awakening, emergence following anesthesia is usually rapid even following more prolonged infusions (>3 hours).
Analogous to the barbiturates, children require higher induction and maintenance doses of propofol on a milligram per kilogram basis as a result of their larger central distribution volume and higher clearance rate. Elderly patients and those in poor health require lower induction and maintenance doses of
propofol as a result of their smaller central distribution volume and decreased clearance rate. Although subhypnotic doses of propofol produce sedation and amnesia,41 awareness has been reported even at higher infusion rates when propofol is used as the sole anesthetic.42 Propofol often produces a subjective feeling of well-being (and even euphoria) on emergence, and it has been abused by health care professionals and others as a result of this CNS action.43
propofol as a result of their smaller central distribution volume and decreased clearance rate. Although subhypnotic doses of propofol produce sedation and amnesia,41 awareness has been reported even at higher infusion rates when propofol is used as the sole anesthetic.42 Propofol often produces a subjective feeling of well-being (and even euphoria) on emergence, and it has been abused by health care professionals and others as a result of this CNS action.43
Propofol produces cortical EEG changes that are similar to those of thiopental. However, sedative doses of propofol increase β-wave activity analogous to the benzodiazepines. Induction of anesthesia with propofol is occasionally accompanied by excitatory motor activity (so-called nonepileptic myoclonia). In a study involving patients without a history of seizure disorders, excitatory movements following propofol were not associated with EEG seizure activity.47 Propofol appears to possess profound anticonvulsant properties.48 Propofol has been reported to decrease spike activity in patients with cortical electrodes implanted for resection of epileptogenic foci and has been used successfully to terminate status epilepticus. The duration of motor and EEG seizure activity following ECT is significantly shorter with propofol than with other IV anesthetics. Propofol produces a decrease in the early components of somatosensory and motor-evoked potentials but does not influence the early components of the auditory-evoked potentials.
Propofol produces dose-dependent respiratory depression, with apnea occurring in 25% to 35% of patients after a typical induction dose. A maintenance infusion of propofol decreases tidal volume and increases respiratory rate. The ventilatory response to carbon dioxide and hypoxia is also significantly decreased by propofol. Propofol can produce bronchodilation in patients with chronic obstructive pulmonary disease and does not inhibit hypoxic pulmonary vasoconstriction.
The cardiovascular depressant effects of propofol are generally considered to be more profound than those of thiopental. Both direct myocardial depressant effects and decreased systemic vascular resistance have been implicated as important factors in producing cardiovascular depression. Direct myocardial depression and peripheral vasodilation are dose- and concentration-dependent. In addition to arterial vasodilation, propofol produces venodilation (caused both to a reduction in sympathetic activity and by a direct effect on the vascular smooth muscle), which further contributes to its hypotensive effect. The relaxation of the vascular smooth muscle may be because of an effect on intracellular calcium mobilization or because of an increase in the production of nitric oxide. Experiments in isolated myocardium suggest that the negative inotropic effect of propofol results from a decrease in intracellular calcium availability secondary to inhibition of transsarcolemmal calcium influx. A recent study in rats has suggested that an altered renin gene is responsible for enhanced sensitivity and hypotensive responses to propofol.49
Propofol also alters the baroreflex mechanism, resulting in a smaller increase in heart rate for a given decrease in arterial pressure.50 The smaller increase in heart rate with propofol may account for the larger decrease in arterial pressure than with an equipotent dose of thiopental. Recent data suggest that induction of anesthesia with propofol attenuates desflurane-mediated sympathetic activation.51 Age enhances the cardiodepressant response to propofol and a reduced dosage is required in studies of the elderly. Patients with limited cardiac reserve seem to tolerate the cardiac depression and systemic vasodilation produced by carefully titrated doses of propofol, and maintenance infusions are increasingly used at the end of cardiac surgery when early extubation is desired.
Propofol does not trigger malignant hyperthermia and may be considered the induction agent of choice in malignant hyperthermia-susceptible patients. The use of propofol infusions for sedation in the pediatric ICU has been linked to several deaths following prolonged administration because of lipid accumulation and hypotension. Although clinical doses of propofol do not affect cortisol synthesis or the response to adrenocorticotropic hormone stimulation, propofol has been reported to inhibit phagocytosis and killing of bacteria in vitro and to reduce proliferative responses when added to lymphocytes from critically ill patients.53 Because fat emulsions are known to support the growth of microorganisms, contamination can occur as a result of dilution or fractionated use.54
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


