Rare Coexisting Diseases
Stephen F. Dierdorf
J. Scott Walton
Andrew F. Stasic
Key Points
Related Matter
Duchenne Muscular Dystrophy
Dantrolene Mixing
Removing Anesthetics from the Breathing Circuit
Raynauds
Scleroderma
Epidermolysis Bullosa
Pemphigus
Rare disorders may influence the conduct of anesthesia. Although the anesthesiologist may encounter such diseases only on rare occasions, he/she must recognize potential interactions between the disease and anesthesia. Advances in molecular medicine continue to clarify disease mechanisms and provide the basis for new therapies.
Musculoskeletal Diseases
The cytoskeleton of the muscle membrane protects the integrity of the membrane and is composed of proteins such as dystrophin, merosin, utrophin, syntrophin, dystrobrevin, and sarcoglycans (Fig. 23-1). Insufficient quantities of normal proteins or the presence of abnormal proteins may weaken the membrane and make it more susceptible to damage.
Muscular Dystrophy
The muscular dystrophies are diseases associated with abnormalities of the muscle membrane and are characterized by progressive loss of skeletal muscle function (Table 23-1) (Fig. 23-2).1 Dysfunctions of cardiac and smooth muscle are less evident but contribute significantly to morbidity and mortality.
Duchenne Muscular Dystrophy
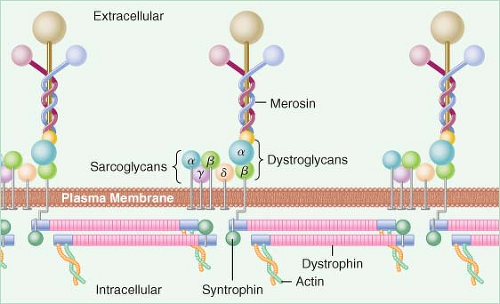 Figure 23.1. Muscle cell cytoskeleton. (Reprinted from: Duggan DJ, Gorospe JR, Fanin M, et al. Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med. 1997;336:618–624, with permission.) |
Table 23-1. Types of Muscular Dystrophy | |||||||
|---|---|---|---|---|---|---|---|
|
Degeneration of cardiac muscle is evidenced by a progressive decrease in R-wave amplitude in the lateral precordial leads of the electrocardiogram. Serial echocardiograms provide important information about cardiac function. Loss of myocardial tissue results in cardiomyopathy, ventricular dysrhythmias, and mitral regurgitation. Treatment of cardiac dysfunction includes angiotensin-converting enzyme (ACE) inhibitors, β-adrenergic blockers, and dysrhythmia surveillance.2
Ineffective cough caused by diminished muscle strength results in retention of pulmonary secretions and pneumonia. Smooth muscle involvement causes intestinal hypomotility, gastroparesis, and delayed gastric emptying.
Although the genetic defect that causes DMD is known, specific gene therapy remains elusive. Current treatment is supportive and directed at improvement of cardiorespiratory function.
Becker Muscular Dystrophy
Dystrophin is present in patients with Becker muscular dystrophy (BMD) but is reduced in amount. The clinical course with respect to skeletal muscle function is milder in BMD patients when compared to patients with DMD. Cardiac involvement is, however, more prevalent in patients with BMD. Patients with BMD may also have epilepsy, macroglossia, and color blindness.3 Female carriers of both DMD and BMD may also have cardiac abnormalities.
Emery–Dreifuss Muscular Dystrophy
Emery–Dreifuss muscular dystrophy is characterized by contractures of the elbow, ankles, spine, and humeropectoral weakness.
The skeletal muscle manifestations are usually mild, whereas cardiac conduction defects can be fatal. Implantable defibrillating cardiac pacemakers are often indicated.
The skeletal muscle manifestations are usually mild, whereas cardiac conduction defects can be fatal. Implantable defibrillating cardiac pacemakers are often indicated.
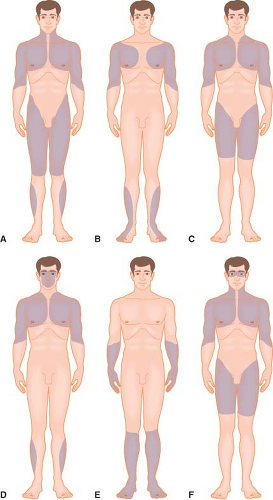 Figure 23.2. Distribution of predominant muscle weakness in different types of muscular dystrophy. (A) Duchenne-type and Becker-type. (B) Emery–Dreifuss. (C) Limb-girdle. (D) Facioscapulohumeral. (E) Distal. (F) Oculopharyngeal. (Reproduced from: BMJ Publishing Group. Emery AE. The muscular dystrophies. BMJ. 1998;317:991–995.) |
Limb-girdle Muscular Dystrophy
Patients with limb-girdle muscular dystrophy exhibit weakness of the shoulder and pelvic girdles. Numerous genetic defects have been discovered that cause abnormalities in different muscle membrane proteins (e.g., calpain-3, dysferlin, and sarcoglycans). Cardiomyopathy and atrioventricular conduction defects can occur.
Facioscapulohumeral Muscular Dystrophy
Patients with this disease have diverse clinical manifestations such as weakness of the facial, scapulohumeral, anterior tibial, and pelvic girdle muscles. Associated abnormalities include retinal vascular disease, deafness, and neurologic dysfunction. Cardiac conduction defects and dysrhythmias may occur.
Oculopharyngeal Muscular Dystrophy
Oculopharyngeal muscular dystrophy (OPMD) presents in late adulthood with ptosis and dysphagia. Dysphagia is secondary to pharyngeal and esophageal muscle weakness. Weakness of the head, neck, and arms may also occur. Patients often require levator resection for ptosis or cricopharyngeal myotomy for dysphagia. Reported anesthesia experience for patients with OPMD is limited. Onset time for non-depolarizing muscle relaxants may be delayed although recovery appears to be normal.4
Congenital Muscular Dystrophy
Congenital muscular dystrophy is characterized by early onset (infancy) of hypotonia, developmental delay, feeding difficulties, and respiratory dysfunction. Included in this group of muscular dystrophies are merosin-deficient muscular dystrophy, Walker–Warburg syndrome, Ulrich disease, muscle–eye–brain disease, rigid spine muscular dystrophy, central core disease, myotubular dystrophy, and nemaline myopathy. Although respiratory dysfunction can be severe, cardiac involvement is not as prominent as in other muscular dystrophies.
Management of Anesthesia
Degeneration of gastrointestinal smooth muscle with hypomotility of the intestine and delayed gastric emptying in conjunction with impaired swallowing increases the risk of perioperative aspiration of gastric contents. Vigorous respiratory therapy and mechanical ventilation may be required after surgery.
The Myotonias
Myotonia is the delayed relaxation of skeletal muscle after voluntary contraction. Electromyography demonstrates repetitive
muscle fiber discharges that fluctuate. These abnormalities are caused by dysfunction of ion channels in the muscle membrane. There are two types of myotonic dystrophy caused by mutations in two distinct gene loci: Myotonic dystrophy type 1 and myotonic dystrophy type 2. The genetic alteration in type 1 is an unstable trinucleotide expansion (CTG) on chromosome 19q. Type 2 is caused by a quadnucleotide expansion (CCTG) on chromosome 3q. Both mutations produce RNA toxicity.
muscle fiber discharges that fluctuate. These abnormalities are caused by dysfunction of ion channels in the muscle membrane. There are two types of myotonic dystrophy caused by mutations in two distinct gene loci: Myotonic dystrophy type 1 and myotonic dystrophy type 2. The genetic alteration in type 1 is an unstable trinucleotide expansion (CTG) on chromosome 19q. Type 2 is caused by a quadnucleotide expansion (CCTG) on chromosome 3q. Both mutations produce RNA toxicity.
Table 23-2. Classification of Myotonic Dystrophy | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
Myotonic dystrophy type 1 is the more common form and is subdivided by age of onset (Table 23-2). Type 1 is a multisystem disease that affects the musculoskeletal system, the heart, the respiratory system, central nervous system, and endocrine system.8 Muscle weakness begins distally and progresses to proximal muscles and muscle wasting occurs over time. Pulmonary function studies demonstrate a restrictive pattern, mild arterial hypoxemia, and diminished ventilatory responses to hypoxia and hypercapnia. Respiratory muscle weakness diminishes the effectiveness of cough and may lead to pneumonia. Aspiration of gastric contents may occur because of gastric atony and pharyngeal muscle dysfunction. Myotonia of the respiratory muscles can produce intense dyspnea.
Other clinical features include cataracts, premature balding, diabetes mellitus, thyroid dysfunction, adrenal insufficiency, and gonadal atrophy. Pregnancy often produces an exacerbation of myotonic dystrophy and congestive heart failure is more likely to occur during pregnancy. Cesarean section is often required because of uterine smooth muscle dysfunction. Infants of mothers with myotonic dystrophy may develop congenital myotonic dystrophy that features hypotonia, feeding difficulty, and respiratory failure.
Until specific molecular therapies are available, treatment is directed at symptom management, such as pacemaker implantation for cardiac dysrhythmias or surgery for cataracts or gallbladder dysfunction.
Management of Anesthesia
Patients with type 1 myotonic dystrophy have far more perioperative complications than patients with type 2 myotonic dystrophy.10,11 Considerations for anesthesia for patients with myotonic dystrophy include the presence of cardiac and respiratory muscle disease and abnormal responses to drugs used during anesthesia. Succinylcholine produces an exaggerated contracture and its use should be avoided (Fig. 23-3). The myotonic response to succinylcholine can be so severe that ventilation and tracheal intubation are difficult. Most patients with type 1 myotonic dystrophy develop a chronic myopathy and the response to nondepolarizing muscle relaxants may be enhanced. Reversal with neostigmine may provoke myotonia. The response to a peripheral nerve stimulator must be carefully evaluated because muscle stimulation may produce myotonia that could be misinterpreted as sustained tetanus when significant neuromuscular blockade still exists. Reversal of rocuronium with sugammadex would be an attractive alternative.
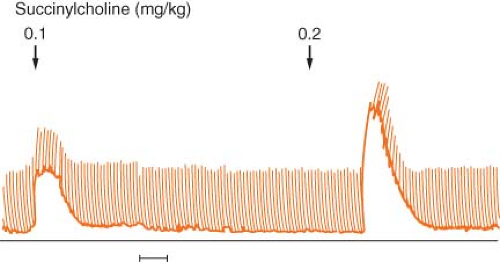 Figure 23.3. Administration of low doses of succinylcholine to a patient with myotonic dystrophy produces an exaggerated contraction of skeletal muscle. (Reprinted from: Mitchell MM, Ali HH, Savarese JJ. Myotonia and neuromuscular blocking drugs. Anesthesiology. 1978;49:44–48, with permission). |
Patients with myotonic dystrophy are sensitive to the respiratory depressant effects of opioids, barbiturates, benzodiazepines, and inhaled anesthetics. Respiratory complications are more likely to occur in the early postoperative period after upper abdominal surgery or in those patients in whom preoperative upper extremity weakness was evident.12
No specific anesthetic technique has been shown to be superior for patients with myotonic dystrophy. Carefully controlled propofol infusions have been used successfully. Inhaled anesthetics may be used but close monitoring of cardiac rhythm and function is indicated. Postoperative mechanical ventilation should be employed until muscle strength and function return.13 Regional anesthesia has been described for both children and adults with myotonic dystrophy.14
Skeletal muscle weakness and myotonia are exacerbated during pregnancy. Labor is typically prolonged and there is an increased incidence of postpartum hemorrhage from placenta accreta. Spinal and epidural anesthesia have been successfully used for pregnant patients.
Familial Periodic Paralysis
The familial periodic paralyses are a subgroup of diseases referred to as the hereditary skeletal muscle ion channelopathies. This group of diseases includes hyperkalemic and hypokalemic periodic paralysis, myotonia congenita, paramyotonia congenita, and the sodium channel myotonias. Alterations in the sodium, potassium, and calcium channels result in muscle membrane inexcitability and weakness.15
Table 23-3. Clinical Features of Familial Periodic Paralysis | |
|---|---|
|
Hyperkalemic Periodic Paralysis
Hyperkalemic periodic paralysis is characterized by episodes of myotonia and muscle weakness that may last for several hours. Weakness can occur during rest after strenuous exercise, infusion of potassium, metabolic acidosis, or hypothermia (Table 23-3). The hyperkalemia is transient and occurs only at the time of weakness. Acute attacks can be fatal because of cardiac dysrhythmias or respiratory failure. A chronic myopathy frequently develops in older patients. Treatment consists of a low potassium diet and the administration of thiazide diuretics.
Hypokalemic Periodic Paralysis
Hypokalemic periodic paralysis is caused by a mutation in two skeletal muscle membrane cation channels. Paralysis can be produced by a decrease in serum potassium levels caused by strenuous exercise, infusion of glucose and insulin, or ingestion of carbohydrates and sodium rich food. Paralysis usually affects the limbs and trunk, but spares the diaphragm. Chronic muscle weakness occurs in most patients as they age.
Thyrotoxic hypokalemic periodic paralysis occurs with thyrotoxicosis in combination with hypokalemia. There is only rarely a family history and presentation is generally later in life. Treatment is directed at treatment of the hyperthyroidism.16
Management of Anesthesia
The primary goal with both forms of familial periodic paralysis is maintenance of normal potassium levels and avoidance of events that precipitate weakness. Any electrolyte abnormality should be corrected prior to surgery. These patients may be sensitive to nondepolarizing muscle relaxants, and short-acting muscle relaxants are preferred. Succinylcholine is best avoided as its administration may alter potassium levels. Metabolic changes (acidosis and alkalosis) or medications (glucose and insulin, diuretics) that reduce potassium levels may cause weakness or paralysis. Because changes in potassium levels precede the onset of weakness, serial measurement of potassium levels during prolonged surgical procedures and the early postoperative period should be considered. The ECG should be monitored for evidence of potassium-related dysrhythmias. Other recommendations include avoidance of carbohydrate loads, hypothermia, and excessive hyperventilation. Halogenated inhaled anesthetics and regional anesthesia have been administered without complication.
Myasthenia Gravis
Myasthenia gravis (MG) is an autoimmune disease with antibodies directed against acetylcholine receptors or other proteins in the postsynaptic membrane of the neuromuscular junction (NMJ). Eighty-five percent of patients with MG have identifiable antiacetylcholine receptor antibodies. Seventy percent of MG patients without AChR (AChR-MG) antibodies have autoantibodies against other NMJ proteins such as muscle specific tyrosine kinase (MuSK, MuSK-MG). It is likely that seronegative MG patients have low antibody titer levels that are not measurable by current assay techniques. The autoantibodies damage the muscle membrane by activation of complement, lysis of the postsynaptic membrane, and loss of postsynaptic folds.17 The thymus may play a central role in the pathogenesis of MG as 90% of MG patients have histologic abnormalities such as thymoma, thymic hyperplasia, or thymic atrophy.
The clinical hallmark of MG is skeletal muscle weakness. The weakness is aggravated by repetitive muscle use and there are periods of exacerbation alternating with remission. Any skeletal muscle can be affected, although there is a predilection for muscles innervated by cranial nerves. Initial symptoms include diplopia, dysarthria, dysphagia, or limb muscle weakness. Myasthenic crises occur in 20% to 30% of MG patients and can be precipitated by poor control of MG, emotional stress, hyperthermia, or pulmonary infections. Myasthenic crisis is characterized by severe muscle weakness and respiratory failure. Cardiac manifestations of MG include focal myocarditis, atrial fibrillation, atrioventricular conduction delay, and left ventricular diastolic dysfunction.
Some pregnant patients have a remission during pregnancy while others (20% to 40%) have increased symptoms during gestation and postpartum respiratory failure can occur. Fifteen percent to twenty percent of neonates born to myasthenic mothers have transient myasthenia from passive transfer of acetylcholine receptor antibodies. Neonatal myasthenia begins 12 to 48 hours after birth and may persist for weeks. Babies from mothers with MuSK-MG have more severe neonatal myasthenia.
Disease classification is based on skeletal muscle groups affected as well as age of onset (Table 23-4). The Osserman staging system is based on the severity of the disease (Table 23-5).
No single test is definitive for the diagnosis of MG and the diagnosis is not always obvious. Serologic testing may detect anti-AChR or anti-MuSK antibodies. Single fiber electromyography is the most sensitive of the electrodiagnostic tests.18
Treatment includes the administration of cholinesterase inhibitors (pyridostigmine), corticosteroids, immunosuppressants, intravenous immunoglobulin (IVG), and plasmapheresis. Cholinesterase inhibitors increase the concentration of acetylcholine (ACh) at the postsynaptic membrane. Consistent control of MG with only pyridostigmine can, however, be quite challenging. Under-dosing will result in residual muscle weakness and over-dosing may produce a “cholinergic crisis” characterized by abdominal pain, salivation, bradycardia, and skeletal muscle weakness. Corticosteroids (prednisone) are nonspecific immunosuppressants used when pyridostigmine does not produce satisfactory control. Azathioprine is the most frequently prescribed
specific immunosuppressant for the treatment of MG. Other immunosuppressants under investigation include cyclosporine, tacrolimus, mycophenolate, cyclophosphamide, methotrexate, and rituximib. The administration of IVG and plasmapheresis are used when rapid treatment is required.19
specific immunosuppressant for the treatment of MG. Other immunosuppressants under investigation include cyclosporine, tacrolimus, mycophenolate, cyclophosphamide, methotrexate, and rituximib. The administration of IVG and plasmapheresis are used when rapid treatment is required.19
Table 23-4. Different Presentations of Myasthenia Gravis | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
The role of thymectomy for the treatment of MG remains controversial. Ten percent of MG patients have a thymoma and thymectomy is clearly indicated. Less invasive surgical techniques for thymectomy such as video-assisted thorascopic surgery (VATS) produce good results with a low incidence of perioperative complications.20
Management of Anesthesia
Table 23-5. Osserman Staging System for Myasthenia Gravis | |
|---|---|
|
Exacerbations of MG should be anticipated during pregnancy. Epidural analgesia can be used during labor and delivery.23 Amide local anesthetics may be better than ester local anesthetics as the metabolism of amides is not affected by cholinesterase activity.
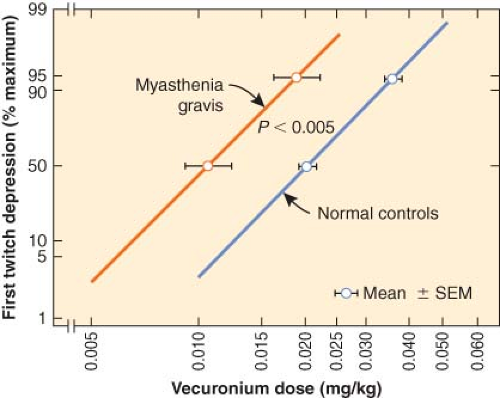 Figure 23.4. Dose-response for vecuronium in normal patients and patients with myasthenia gravis. (Reprinted from: Eisenkraft JB, Book WJ, Papatestas AE. Sensitivity to vecuronium in myasthenia gravis: A dose-response study. Can J Anaesth. 1990;37:301–306, with permission.) |
Table 23-6. Comparison of Myasthenic Syndrome and Myasthenia Gravis | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Myasthenic Syndrome (Lambert–Eaton)
Treatment of the underlying neoplasm may improve the neurologic condition. The most effective drug for the treatment of LEMS associated muscle weakness is 3,4-diaminopyridine. 3,4-diaminopyridine prolongs the presynaptic action potential and increases the release of acetylcholine. Immunosuppression with corticosteroids and azathioprine may also be beneficial. Plasmapheresis and intravenous immunoglobulin may produce short-term improvement.25
Management of Anesthesia
Patients with LEMS are sensitive to the effects of both depolarizing and non-depolarizing muscle relaxants. The administration of 3,4-diaminopyridine should be continued until the time of surgery. The possibility of occult muscle weakness should be considered in any patient with a malignant tumor.
Guillain–Barré Syndrome (Polyradiculoneuritis)
Guillain–Barré syndrome (GBS) is the acute form of a group of disorders classified as the inflammatory neuropathies. Other diseases in this group include acute inflammatory demyelinating polyneuropathy (AIDP), acute motor axonal neuropathy (AMAN), acute motor-sensory axonal neuropathy, Miller-Fisher syndrome, and chronic inflammatory demyelinating polyneuropathy (CIDP).
GBS is an autoimmune disease triggered by a viral or bacterial infection and is an example of molecular mimicry and cross-reactivity. The infectious agent produces a substance that causes an immune reaction. Unfortunately, that substance resembles a neural component of the host and autoantibodies develop that attack the host. Patients with GBS develop antibodies to gangliosides in the peripheral nerves.26 Most patients with GBS have a history of a respiratory or gastrointestinal infection within 4 weeks of the onset of neurologic symptoms. Infections with Campylobacter jejuni, Haemophilus influenzae, Mycoplasma pneumoniae, Epstein-Barr virus, and cytomegalovirus are most frequently associated with GBS. There are sporadic case reports of GBS or a GBS-like syndrome presenting after surgical procedures.
GBS is characterized by onset of skeletal muscle weakness or paralysis of the legs. Paresthesias may precede the weakness. The paralysis progresses cephalad to include muscles of the trunk and arms with maximal weakness developing 2 to 4 weeks after the onset. A plateau phase usually develops before recovery begins. The most serious problem is ventilatory insufficiency and 25% of patients with GBS will require mechanical ventilation. Although 85% of GBS patients achieve a good recovery, 3% to 5% develop a chronic, recurrent neuropathy.
Autonomic nervous system dysfunction can produce wide fluctuations in cardiovascular parameters. In a manner similar to autonomic hyperreflexia, physical stimulation can precipitate hypertension, tachycardia, and cardiac dysrhythmias.
Plasma exchange and the administration of intravenous immunoglobulin have been shown to be effective for patients with GBS.27
Management of Anesthesia
Autonomic nervous system dysfunction may cause hypotension secondary to postural changes, blood loss, or positive pressure ventilation. Noxious stimuli such as laryngoscopy and tracheal intubation may produce exaggerated increases in heart rate and blood pressure.
Succinylcholine should be avoided because of the danger of hyperkalemia. This risk may persist after clinical recovery from GBS.28 A short-acting non-depolarizing muscle relaxant with minimal cardiovascular effects (cisatracurium, rocuronium) would be a useful choice. The sensitivity to non-depolarizing muscle relaxants may vary from extreme sensitivity to resistance, depending on
the phase of the disease.29 It is likely that mechanical ventilation may be required in the immediate postoperative period. Patients with GBS who have pronounced sensory disturbances may benefit from neuraxial opioids.
the phase of the disease.29 It is likely that mechanical ventilation may be required in the immediate postoperative period. Patients with GBS who have pronounced sensory disturbances may benefit from neuraxial opioids.
Central Nervous System Diseases
Multiple Sclerosis
The features of multiple sclerosis (MS) are inflammation and demyelination in the brain and spinal cord. Although circulating T cells cannot normally penetrate the blood–brain barrier, an environmental stimulus (virus) may activate T cells. The activated T cells cross the blood–brain barrier and initiate an inflammatory and immunologic attack on myelin.30,31 Demyelination interferes with neural transmission and causes CNS dysfunction. The ability of neural tissue to repair itself during the early phases of the disease explains the relapsing nature of MS. There is also evidence that damage is not restricted to white matter and that gray matter is also affected.
The symptoms of MS depend on the sites of demyelination. Brainstem involvement can produce nystagmus, diplopia, trigeminal neuralgia, autonomic dysfunction, and alterations in ventilation that lead to hypoxemia and respiratory failure. Lesions of the spinal cord produce weakness and paresthesias. The legs are affected more than the arms. Bowel retention and urinary incontinence are frequent complaints. The course of MS is characterized by exacerbations of symptoms at unpredictable intervals over a period of years. Patients are classified during the early phases as either relapsing–remitting (85%) or primary progressive MS. Many patients with relapsing–remitting MS develop neurodegeneration and are categorized as secondary progressive MS. Patients with primary progressive MS are devoid of acute episodes, but develop progressive neurodegeneration. Pregnancy is associated with improvement of symptoms, but relapse often occurs within the first three postpartum months.
Clinical criteria for the diagnosis of MS include onset between 10 and 50 years of age, signs and symptoms of CNS white matter disease, two or more attacks separated by a month or more, and involvement of two or more non-contiguous anatomic areas. Elevated levels of IgG and albumin in the cerebrospinal fluid are characteristic of MS. Magnetic resonance imaging (MRI) is a sensitive diagnostic tool for MS and provides direct evidence of the location of demyelinated plaques in the CNS.32
Therapy for MS is directed at modulating the immunologic and inflammatory responses that damage the CNS. Current therapy for MS includes interferon preparations, glatiramer, mitoxantrone, and monoclonal antibodies. Interferon works by reducing T cell transit across the blood–brain barrier. Glatiramer is a mixture of polypeptides that mimic the structure of myelin and serve as a decoy for autoantibodies. Mitoxantrone is related to the antineoplastic drugs doxorubicin and may be cardiotoxic. Natalizumab prevents T cell migration but can produce a fatal leukoencephalopathy. The most recent therapeutic breakthrough in the treatment of MS has been the development of oral agents.33 These include cladribine, fingolimod, laquinimod, teriflunomide, and dimethyl fumarate. Corticosteroids are still used for acute relapses that do not respond to other medications.
Many patients with MS experience chronic pain, including central neuropathic pain, paresthesias, trigeminal neuralgia, and spasticity. Symptomatic therapy includes diazepam, dantrolene, and baclofen for spasticity. Painful dysesthesias, tonic seizures, and ataxia can be treated with carbamazepine. Nonspecific measures include the avoidance of excessive fatigue, emotional stress, and hyperthermia. Demyelinated fibers are extremely sensitive to increases in temperature and hyperthermia can block impulse conduction.
Management of Anesthesia
The effect of surgery and anesthesia on the course of MS is controversial. Some reports have indicated that regional anesthesia and general anesthesia have been reported to exacerbate MS. Other reports, however, have found no correlation between anesthesia and the course of the disease. Factors other than anesthesia such as infection, hyperpyrexia, and emotional stress may contribute to an increased risk of an exacerbation. Preoperatively, the patient should be advised that surgery and anesthesia could produce a relapse despite a well-managed anesthetic.
Autonomic dysfunction may enhance the hypotensive effects of volatile anesthetics. Drug interactions that may be anticipated during the perioperative period include cardiotoxicity from immunosuppressants, and altered responses to muscle relaxants. Baclofen increases the sensitivity to nondepolarizing muscle relaxants and anticonvulsants produce resistance to nondepolarizing muscle relaxants. Succinylcholine could produce an exaggerated release of potassium. Respiratory muscle weakness and respiratory control dysfunction increase the likelihood of respiratory support during the immediate postoperative period.35
Epilepsy
A seizure is a common manifestation of many types of CNS disease and is the external manifestation of epilepsy. A seizure results from the excessive discharge of large numbers of neurons that become depolarized in a synchronous fashion. Epilepsy (idiopathic seizures) begins in childhood. The sudden onset of seizures in an adult may indicate focal brain disease (tumor). The onset of seizures after 60 years of age can be a result of cerebrovascular disease, head injury, tumor, infection, or a metabolic disturbance.
Although there are many antiseizure drugs, 30% of epileptic patients are poorly controlled and sudden death may occur in adults with epilepsy since childhood (Table 23-7).36,37 The newer antiseizure drugs target ion channels, GABA receptors, amino acid receptors, and synaptic proteins.38
The most frequently encountered types of seizures are:
Grand mal seizure: A grand mal seizure is characterized by generalized tonic–clonic activity. All respiratory effort is arrested and a period of arterial hypoxemia ensures. The tonic phase lasts 20 to 40 seconds and is followed by the clonic phase. In the postictal period, the patient is lethargic and confused. Diazepam, thiopental, and propofol are effective for treatment of acute, generalized seizures. Epileptic patients resistant to drug therapy may benefit from surgical resection of a seizure focus or implantation of a vagal nerve stimulator (VNS).
Focal cortical seizure: Focal cortical seizures may be motor or sensory depending on the site of neuronal discharge. There
is usually no loss of consciousness, although the focal seizure may induce a grand mal seizure.
Table 23-7. Antiepileptic Drugs (AED) Classified by Mechanism of Action
Enhance GABAergic System
Barbiturates
Benzodiazepines
Primidone
Tiagibine
Vigabatrin
Affect Cation Channels
Phenytoin
Carbamazepine
Oxcarbazepine
Lamotrigine
Topiramate
Ethosuximide
Valproate
Retigabine
Zonisamide
Antagonists of Excitatory Amino Acids
Felbamate
Lacosamide
Talampanel
Unknown Mechanism of Action
Acetazolamide
Sulthiame
Gabapentin
Pregabalin
Levetiracetam
AEDs for Status Epilepticus
Propofol
Diazepam
Lorazepam
Midazolam
Fosphenytoin
Absence seizure (petit mal): Absence seizures are characterized by a brief loss of awareness (30 seconds). Additional manifestations include staring, blinking, and rolling of the eyes. Absence seizures typically occur in children and young adults.
Akinetic seizure: Akinetic seizures are a sudden, brief loss of consciousness and postural tone. These types of seizures usually occur in children and can result in a severe head injury from the fall.
Status epilepticus: Status epilepticus is defined as two consecutive tonic–clonic seizures without regaining consciousness, or seizure activity that is unabated for 30 minutes or more. Grand mal status epilepticus may last for more than 48 hours with a seizure frequency of 4 to 5 per hour and mortality can be as high as 20%. As the seizure progresses, skeletal muscle activity diminishes and seizure activity may only be evident on the EEG. Respiratory effects of status epilepticus include inhibition of respiratory centers, uncoordinated skeletal muscle activity that impairs ventilation and bronchoconstriction. Phenobarbital, phenytoin, benzodiazepines, thiopental, and propofol are good choices for initial therapy. On rare occasions, general anesthesia may be required.
Management of Anesthesia
Most inhaled anesthetics, including nitrous oxide, have been reported to produce seizure activity. Such activity during the administration of isoflurane and desflurane is extremely rare as these drugs produce a dose-dependent depression of EEG activity. Sevoflurane may be epileptogenic, although the clinical significance is uncertain.39 Stimulation of hepatic microsomal enzymes by anticonvulsant drugs may increase the rate of biotransformation of halogenated anesthetics and increase the risk of organ damage. Ketamine may produce seizure activity and it would seem reasonable to avoid its use, as there are other suitable induction agents (e.g., thiopental, propofol, benzodiazepines). Potent opioids such as fentanyl, sufentanil, and remifentanil may produce myoclonic activity or chest wall rigidity that may be confused with seizure activity. Despite these concerns, the degree of preoperative seizure control may be a more important predictor of perioperative seizures.40 Local anesthetics can decrease the seizure threshold but there is no evidence that regional anesthesia increases the risk of seizures.41
Side effects of antiseizure medications include leucopenia, anemia, hepatitis and hepatic failure, pancreatitis, coagulopathy, cardiotoxicity, hypothyroidism, and skin rash.42 Complications of vagal nerve stimulators include vocal cord paralysis, facial palsy, bradycardia/asystole, and airway obstruction.43 Appropriate airway resuscitation equipment and rapid-acting anticonvulsants such as barbiturates, propofol, and benzodiazepines should be readily available if a perioperative seizure occurs.
Alzheimer Disease
Alzheimer disease is the major cause of dementia in the United States and there are over 5 million people in the United States with the disease. The incidence is 5% in persons over the age of 65 years and 30% over the age of 85 years. Alzheimer disease is characterized by cognitive impairment, poor decision making, language deterioration, gait disturbances, seizures, agitation, and psychosis. A positive diagnosis can, however, only be made at autopsy. Imaging studies show hippocampal atrophy (MRI) and glucose hypometabolism (PET). The deposition of amyloid β peptides appears to be central to the degeneration and death of neurons. Deposition of amyloid β peptides produces amyloid plaques and neurofibrillary tangles and activates the apoptotic cell death cascade. There is no specific therapy for Alzheimer disease. Initial symptomatic therapy is with cholinesterase inhibitors (donepezil, rivastigmine, galantamine) and an NMDA receptor antagonist (memantine). Cholinesterase inhibitors improve the patient’s ability to perform daily living activities and may improve cognition. Side effects of cholinesterase inhibitors include nausea, emesis, bradycardia, syncope, and fatigue. Antidepressants, anticonvulsants, and antipsychotics are used for neuropsychiatric symptoms as necessary. A large number of experimental therapies are directed at disease modifying pathways with immunotherapy, secretase inhibitors, amyloid aggregators, chelation, and Tau aggregation inhibitors.44
Management of Anesthesia
Animal studies have demonstrated that volatile, halogenated anesthetics produce neuronal changes that resemble the diseased neurons of patients with Alzheimer disease. These findings have
caused significant controversy about the use of halogenated anesthetics in neonates and elderly patients.45,46,47 Any clinical recommendations concerning the use of halogenated, inhaled anesthetics are, however, speculative. Postoperative cognitive dysfunction (POCD) is well known in elderly patients, but the causes remain elusive. Patients and their families should be advised that POCD can occur.
caused significant controversy about the use of halogenated anesthetics in neonates and elderly patients.45,46,47 Any clinical recommendations concerning the use of halogenated, inhaled anesthetics are, however, speculative. Postoperative cognitive dysfunction (POCD) is well known in elderly patients, but the causes remain elusive. Patients and their families should be advised that POCD can occur.
The selection of anesthetics and techniques will be influenced by the patient’s physiologic condition and the degree of neurologic impairment. The patient’s preoperative drug list should be reviewed for the possibility of interactions with anesthetics. Patients are likely to be confused and uncooperative because of dementia. Sedative premedication should be used with caution, if at all, as mental confusion may worsen. If an anticholinergic is required, glycopyrrolate, which does not cross the blood–brain barrier, is preferable to atropine or scopolamine. Patients receiving cholinesterase inhibitors may have a prolonged response to succinylcholine.
Parkinson Disease
Parkinson disease (PD) is a degenerative CNS disease caused by a loss of dopaminergic cells in the basal ganglia of the brain. The characteristic pathologic feature is the presence of Lewy bodies in the neurons of the substantia nigra. Lewy bodies are aggregations of damaged proteins. The etiology of PD is an interaction of a genetic predisposition and unidentified environmental factors. Other than the well-described postencephalitic PD, there is little evidence that PD is caused by a virus.48
The clinical effects of PD are caused by dopamine deficiency. The most characteristic features of PD are resting tremor, cogwheel rigidity of the extremities, bradykinesia, shuffling gait, stooped posture, and facial immobility. These features are secondary to diminished inhibition of the extrapyramidal motor system as a result of dopamine deficiency. Other clinical features include seborrhea, sialorrhea, orthostatic hypotension, bladder dysfunction, diaphragmatic spasm, oculogyric crisis, dementia, and mental depression.
Treatment is directed toward increasing dopamine levels in the brain, but preventing the adverse peripheral effects of dopamine. Levodopa is the single most effective drug for patients with PD. When administered orally, levodopa is converted to dopamine and causes side effects such as nausea, emesia, and hypotension. To avoid such side effects, levodopa is administered with carbidopa and entacopone. Carbidopa is a peripheral decarboxylase inhibitor and entacopone is a catechol-O-methyltransferase inhibitor that increases the bioavailability of levodopa. Other drugs that improve function are the monoamine oxidase-B inhibitors, selegiline and rasagiline. Dopamine receptor agonists such as bromocriptine, pramipexole, ropinirole, pergolide, and cabergoline can also be used for the treatment of PD. Pergolide and cabergoline are ergot-derived drugs that can cause cardiac valvular fibrosis and insufficiency. Implantation of deep-brain stimulators (DBS) may be quite effective for patients with advanced PD. The therapeutic regimen for patients with PD is complex and requires a skilled neurologist to individualize therapy.49
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


