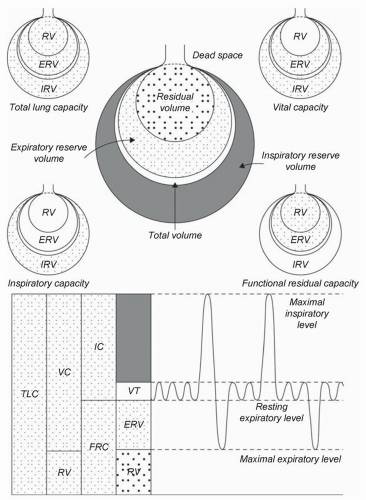
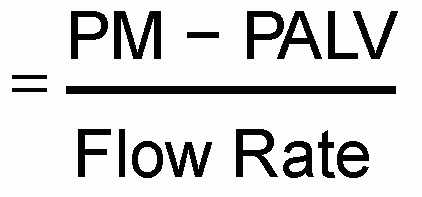Figure 6.1 is a graphic representation of lung volume components that are meaningful for both negative pressure and positive pressure respiration (
1). Tidal volume (VT) is the most frequent volume of air moved into and out of the lungs. Functional residual capacity (FRC), the amount of volume remaining in the lung after a normal expiration, is particularly relevant to upcoming discussions of respiratory failure and ventilator management. Critical closing volume (CCV, not shown in
Fig. 6.1) is the lung volume at which small airways collapse, resulting in microat-electasis. Normally, CCV is less than FRC. However, with increasing age, chronic lung disease, and acute lung disease, CCV may become larger than FRC, resulting in significant atelectasis and an increase in physiologic shunt fraction (
2).
Determinants of Arterial PCO2 and PO2
The determinants of arterial PCO
2 and PO
2 (PaCO
2, PaO
2) are listed in
Table 6.1. PCO
2 is directly proportional to the ratio of dead space to tidal volume, VD/VT. As VD ventilation approaches VT, carbon dioxide cannot be eliminated (VD/VT = 1). Methods used to reduce an elevated PCO
2 include reduction of VD (i.e., tracheostomy to replace an endotracheal tube) or an increase in VT (i.e., relief of bronchospasm). Increasing rate of respiration to lower PCO
2 will be successful only if there are areas of lung having a low VD/VT ratio. If the entire lung has a high VD/VT ratio, simply increasing respiratory rate will be ineffective (
3).
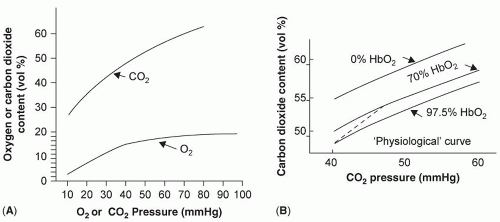
The carboxyhemoglobin dissociation curve is steep and almost linear, as compared to the oxyhemoglobin dissociation curve (
Fig. 6.3). Increasing oxygen saturation reduces affinity and facilitates CO
2 release in the lungs; lower oxygen saturation increases affinity and augments CO
2 removal from the tissues. The nature of this curve allows a well ventilated and perfused area of lung to markedly reduce CO
2 content and compensate for lung areas not engaged in gas exchange (
1).
Carbon dioxide is the end product of the metabolism of carbohydrate, protein, and fat. Increased metabolic demands (e.g., exercise, infection) increase O2 use and CO2 production. The ratio of CO2 production to O2 use is respiratory quotient (RQ), which is 1.0 for carbohydrate and protein and 0.7 for fat. Hypermetabolic states that use primarily carbohydrate and protein (e.g., severe inflammation) result in more CO2 production than states that use primarily fat (starvation without inflammation).
The alveolar gas equation determines alveolar PO2 (PAO2) and, therefore, the highest possible PaO2.
PAO2 = (FiO2 × (Patmos − PH2O)) − (PACO2 × [FiO2 + (1 − FiO2)/RQ]
Where, FiO
2 is the fraction of oxygen in inspired air, P
atmos is the atmospheric pressure (760 mm Hg at sea level), P
H2O is the partial pressure of water in the alveolus (47 mm Hg at 37°C), P
ACO
2 is mean alveolar PCO
2 (usually close to arterial PCO
2) (
1).
Increasing body temperature and the resultant increase in PH2O as well as elevated PACO2 can both lower PaO2, but the effect will be less as FiO2 increases. The alveolar gas equation is the clearest demonstration of the interaction between oxygen being added to the blood and CO2 being eliminated. Otherwise, the two processes are best thought of as separate.
Ideally, the ventilation and perfusion of alveoli are perfectly matched (V/Q = 1). Areas that are ventilated but not perfused (V/Q = infinity) are physiologic VD units; areas that are perfused but not ventilated (V/Q = 0) are physiologic shunts. A shunt (Qs) represents venous blood that does not come in contact with ventilated alveoli (
Fig. 6.2, middle and bottom). Similar to VD, there is a normal anatomic component of shunting [5-6% of cardiac output (Qt)] that consists principally of the blood supply to the bronchioles and heart, which then drains directly
into the pulmonary veins or the left ventricle, respectively. Areas of lung that simulate VD will affect PaO
2 via the alveolar gas equation. Areas that simulate a shunt will diminish PaO
2 by the admixture of venous blood with newly oxygenated blood.
With minimal shunting, mixed venous oxygen content will have little effect on PaO
2. Increasing the shunt increases this influence. Under these circumstances, variables that decrease mixed venous oxygen saturation (decreased cardiac output, increased oxygen utilization) can significantly diminish PaO
2 and may lead to therapy directed at improving lung function (
4). Similarly, increasing cardiac output may improve PaO
2 and lead to a false interpretation that the lung is better. Measurement of cardiac output and calculation of the shunt percentage as well as oxygen delivery and consumption, can help sort out the influence of cardiac output and oxygen consumption on arterial oxygen concentration.
Shunt equation (percent of cardiac output engaged in the shunt effect):
Qs/Qt = [(PAO2 − PaO2) × 0.0031]/[(CaO2 − CvO2) + (PAO2 − PaO2) × 0.0031]
where CaO
2 is arterial oxygen content and CvO
2 is mixed venous oxygen content (
3).
Oxygen content = 1.34 × hemoglobin (g/dL) × oxygen saturation + 0.0031 × PO2
The average V/Q relationship of a normal lung is close to unity, but gravity is a determinant of the relative ratio of ventilation to perfusion in different areas of the lung. For instance, pulmonary blood flow is several times greater at the bases as compared to the apex in an upright man (
5). Proportionally, however, ventilation is greatest at the apex. West has divided the lung into three zones that describe the ratio of intra-alveolar pressure (PAV) to pulmonary arterial (PA) and venous (PV) pressure: Zone I, PAV > PA > PV; Zone II, PA > PAV > PV; Zone III, PA > PV > PAV (
Fig. 6.4) (
6). Pulmonary artery occlusion pressure equals pulmonary venous pressure and, therefore, measures left atrial pressure most reliably in Zone III, where the catheter tip is vertically below the left atrium. Fortunately, since the catheter is flow directed, most often the tip does locate in Zone III.
More recent investigation has demonstrated that this vertical, gravity-dependent influence on ventilation and perfusion is accompanied by equal, if not greater, differences in the horizontal (isogravitational) planes of the lung. It is possible for high-perfusion regions to persist regardless of posture and blood flow is greater in the central regions of the lung as compared to the periphery. Similar findings have been noted with ventilation (
5).
Diminished diffusion capacity is generally of little clinical significance in surgical critical care and likely to cause hypoxemia only when FiO2 is low (high altitude), with thickened alveolar capillary membranes (interstitial fibrosis), or with shortened exchange time (very high cardiac output).
Compliance/Elastance
Expansion of a lung can be likened to the expansion of a balloon. In a comparison of two balloons subjected to the same increment in transmural pressure, if balloon 1 expands to a larger
volume than balloon 2, then balloon 1 has more compliance but less elastance. Compliance is the ratio of the change in pressure to the change in volume. Elastance is the opposite of compliance and represents the intrinsic elastic component that resists deformation by stress. The relationship between transmural inspiratory and expiratory pressures and lung volumes is determined by the compliance of the chest wall and lungs (total compliance,
Table 6.3). Without disease, total compliance (CT) is determined mostly by the elastic recoil properties of the lung (CL) and thorax (CW). With negative pressure, the inspiration CT equals approximately 0.1 L/cm H
2O. Under conditions of mechanical ventilation CT in patients with normal lungs and chest walls, CT is approximately 0.05 L/cm H
2O. Certain diseases (e.g., circumferential thoracic burns) diminish primarily chest wall compliance, while others (e.g., pulmonary edema) diminish primarily lung compliance. Critically ill patients commonly develop alterations in both components of total compliance.
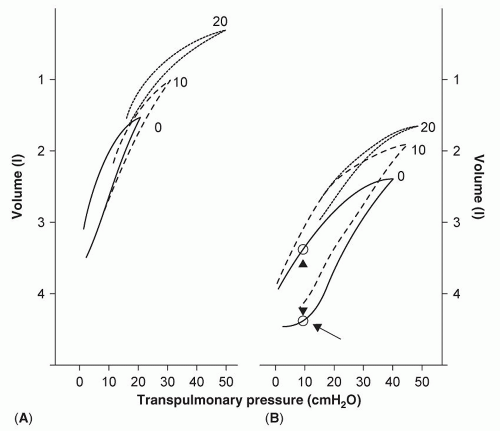
The compliance described above is measured as a given inflation volume is held constant (static compliance). The relationship between volume and pressure can be plotted (pressure-volume curve, P-V curve,
Fig. 6.5) and the slope of this curve represents compliance. As seen in the figure, a normal lung exhibits a similar relationship between pressure and volume as pressure is gradually increased and then decreased (left part of the figure). With certain diseases [acute respiratory distress syndrome (ARDS) in this case, right part of the figure], the pressure volume curve exhibits significant hysteresis, indicating that compliance changes as lung volume units open when pressure is increased and then close as pressure is decreased. The changes in static compliance with different inflation volumes can be plotted (
Fig. 6.6) to produce a characteristically sigmoidal curve. An increase in compliance is greatest in the mid-volume range (where VTs usually occur) and is the least at total lung capacity (top right of the curve) and low lung volumes (bottom left of the curve) near FRC or RV.
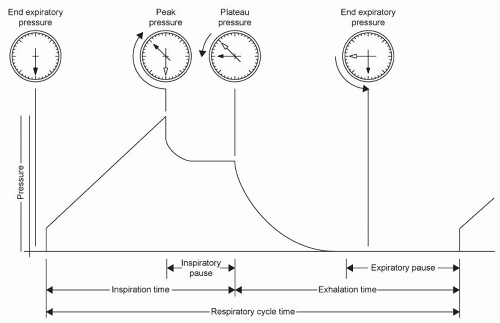
Airway Pressures
During negative pressure ventilation, the lung is subjected to little potential damage from pressure effects. Positive pressure ventilation, however, results in several airway pressure alterations that may yield beneficial effects (improved oxygenation and carbon dioxide removal) and detrimental effects (decreased cardiac output and lung damage). The airway
pressures that have received the most attention in surgical critical care are as follows: mean airway pressure ([P with bar above]aw), peak inspiratory pressure (PIP), the pressure at end inspiration (plateau pressure, Pplat), alveolar pressure (Palv), transpleural pressure (Ptrans), and positive pressure at the end of expiration (PEEP).
[P with bar above]aw is the mean pressure monitored in the airway during the entire respiratory cycle. [P with bar above]aw is influenced by mean alveolar pressure, but is not a direct measurement, being influenced by such variables as inspiratory resistance as well as the inspiratory/expiratory time cycle. Despite this, [P with bar above]aw is a variable that can be linked to the oxygenation and cardiovascular effects of positive pressure mechanical ventilation. On average, as [P with bar above]aw increases, oxygenation improves and venous return decreases (
7).
PIP is the maximum pressure generated in the airway during gas flow (
Fig. 6.7). PIP can be influenced by compliance, airway resistance, VT, and the rate of flow of gas. Pplat is the pressure in the airway at the end of inspiration during a positive pressure VT, but before exhalation begins (inspiratory pause). Pplat
(Fig. 6.7) is a measure of peak alveolar pressure. During the inspiratory pause, there is a drop from PIP as gas distributes from the upper to the lower airways. Pplat is mostly affected by total thoracic compliance (
8).
Palv is the pressure in the alveolus during the entire respiratory cycle. Palv is closely approximated by [P with bar above]aw in many clinical circumstances, especially when inspiratory and expiratory resistances are nearly equal and airway flow rates are low. At zero flow, [P with bar above]aw does equal Palv. However, during flow and especially when expiratory resistance is higher than inspiratory resistance, [P with bar above]aw will underestimate Palv, especially when minute ventilation is high. That is, when air meets more resistance leaving the alveolus than when air is brought to the alveolus, alveolar pressure will increase in proportion to the amount of air brought to the alveolus each minute (
9).
The change of Palv during the respiratory cycle has a direct influence on pleural pressure (Ppl), which is also influenced by the compliance of the lung (CL) and chest wall (CW) (
7).
Δ Ppl = Δ Palv × (CL/(CL + CW))
Ppl is inversely related to alterations in venous return. That is, an increase in Ppl will result in a decrease in venous return. Therefore, an increase in Palv in compliant lungs will
have a more significant effect on Ppl and venous return than an increase in Palv in non-compliant lungs (
7). On the other hand, non-compliant lungs will develop less of an increase in pleural pressure and more of an increase in transpulmonary pressure (Ptrans) as Palv increases.
Ptrans is the pressure difference between the alveolus and the pleural space, as described by the following formula:
As discussed in the section on “Ventilator-Induced Lung Injury,” Ptrans is an important feature of the risk of ventilator-induced lung injury (VILI) (
10).
PEEP is pressure present in the airway at the end of expiration. PEEP is often applied to the ventilator circuit as part of ventilator management (external PEEP). In addition, disease states that result in a failure to return to passive FRC before the onset of the next inspiration (high expiratory resistance − dynamic hyperinflation), and ventilator settings that result in a similar phenomenon (increased inspiratory time to expiratory time ratio – I:E ratio) can result in increased alveolar pressure at the end of expiration (auto-PEEP). For patients on a ventilator, auto-PEEP may be present whenever the flow tracing shows flow at the end of exhalation. Total PEEP is the sum of externally applied PEEP and auto-PEEP. Measurement of end-expiratory occlusion pressure (the airway pressure at end-expiration with the expiratory port occluded) provides total PEEP. The difference between this and externally applied PEEP is auto-PEEP. Auto-PEEP can increase Paw and Palv in the same fashion as external PEEP and, therefore, has the same potential to influence hemodynamics and transalveolar pressure (
8).
Pulmonary Fluid
The physiology of the movement of intravascular fluid and protein into and out of the pulmonary interstitium has been well studied and is described by Starling’s equation:
Jv = Lp × A × [(Pc − Pt) – σ(πc − πt)]
Where Jv is the magnitude of fluid migration per unit of time; Lp is hydraulic conductance or the speed at which fluid can pass through the microvascular exchange barrier; A is the
surface area available for exchange; σ is the reflection coefficient, or the relative permeability of the microvascular membrane to plasma proteins; Pc is the plasma hydrostatic pressure; Pt is the interstitial hydrostatic pressure; πc is the plasma colloid oncotic pressure; πt is the interstitial colloid oncotic pressure (
11,
12).
In the lung, as in other organs, the migration of fluid and protein, especially albumin, into the interstitial space is normal at the high pressure end of capillaries, is partially returned to the circulation as the hydrostatic pressure falls, but also returns to the circulation via the lymphatics. Interstitial and subsequent alveolar edema does not occur until the lymphatics are overwhelmed. Pulmonary lymphatics may be capable of removing several times the usual amount of interstitial fluid before edema develops. Pulmonary lymphatics appear to be more capable of this function than those that drain the systemic circulation (
13,
14,
15).
Starling’s equation and the physiology of lymphatic drainage allow for several etiologies of pulmonary edema (
Table 6.5) (
12). The “true” hydrostatic pressure in the pulmonary capillary (Pcap) is determined by mean PA pressure and LAP or PAOP, as well as pulmonary arterial and venous resistance (see
chap. 3) (
16). While normal conditions allow an estimate of Pcap from a published formula, disease alters the listed variables sufficiently to preclude accuracy (
Table 6.6). Measurement of Pcap is possible using the pulmonary artery tracing available through a pulmonary artery catheter. Disease will usually result in an increase in Pcap compared to PAOP (
16), but the increase is not typically large, (i.e., <5 mm Hg).
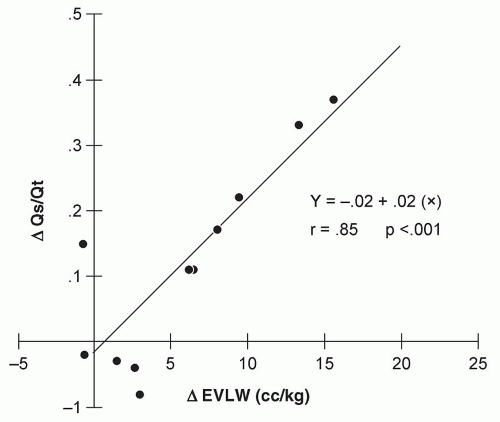
The most common etiology of an increase in Pc is left heart failure. When Pc is elevated, a good correlation between radiographic indices of increased lung water and hydrostatic pressure has been documented (
Table 6.7). In addition, an increase in hydrostatic pressure (typically >20 mm Hg) can result in increased extravascular lung water (EVLW) that correlates well with deficits in oxygenation (
Fig. 6.8) (
16,
17,
18).
More controversial is the effect of low plasma oncotic pressure (πc) with normal hydrostatic pressure and normal pulmonary capillary permeability. Experimental data are conflicting, but human data do not support this as a principal etiology of lung water accumulation (
12,
15,
16,
19,
20).
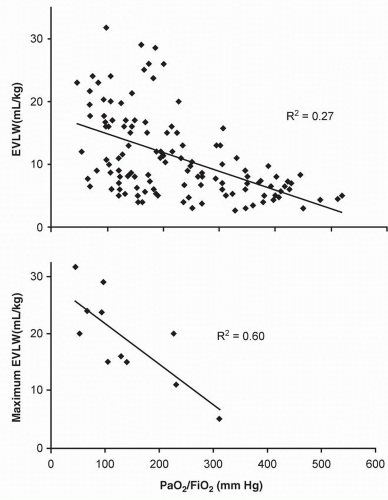
An increase in pulmonary capillary permeability as a mechanism of interstitial and alveolar fluid accumulation has been extensively investigated in both experimental and human models. In contrast to an increase in Pc, an increase in permeability (σ) does not result in a good correlation between EVLW and either radiographic or oxygenation alterations (
Fig. 6.9) (
21,
22,
23,
24,
25,
26,
27,
28,
29). As further discussed in the section “Effects of Systemic Inflammation on the Lung” EVLW contributes, but does not completely explain the pathophysiological alterations characteristic of permeability pulmonary edema. Therefore, management strategies differ from that employed when hydrostatic edema is the principal alteration.
The most common diseases obstructing lymphatics are malignancies that either invade the mediastinal lymphatics (lymphoma, metastatic lung cancer) or spread in the interstitium of the lung (breast cancer, leukemia). This form of pulmonary edema is usually unresponsive to common treatment but may improve if the malignancy is treated.