Fig. 5.1
Distribution of age at death in patients with congenital heart disease in 1987–1988 and 2004–2005. Histogram bars depict the proportion of all deaths (x-axis) according to age at death (y-axis) in our cohort of patients with congenital heart disease in the first (1987–1988; left graph) and final (2004–2005; right graph) years of observation. Bold black curves with diamonds represent the corresponding age at death distribution in the general Quebec population during the same periods of observation (Reproduced from Avila et al. [1])
Demographic changes in this population could be explained by the major and continuous improvements in different areas, starting with prenatal screening for CHD during pregnancy and at birth, alongside with advances in the medical and interventional management of these babies. Tremendous progress has been also achieved in surgical repair, especially in cardiopulmonary bypass techniques, anesthesia, and postoperative management. Currently, there are more adults living with CHD than children. Recent demographic analysis demonstrated that CHD-related death in pediatrics has decreased by 30% between two time periods: 1987–1988 and 2004–2005. It also showed that the decrease was much more noticeable in severe CHD: 67% less mortality [1].
More than 85% of the children born with CHD survive now to adulthood. This phenomenon explains why this cohort tends to grow old. In approximately 20 years, the median age at death increased from 2 to 23 years for complex CHD. For adults with CHD, the median age at death has also shifted from 60 to 75 years. Children with CHD reaching adulthood are estimated to be approximately one million in the United States, 1.8 million in Europe, and around 100,000 in Canada [2, 3].
In a recent study published in 2016, Agarwal assessed the burden of ACHD presenting to US emergency departments [4]. Numbers increased significantly between 2006 and 2012, with a striking increase in patients presenting with PHT. There was an increase in the prevalence of cardiovascular risks among these patients, including smoking, obesity, hypertension, diabetes, and vascular disease, as well as respiratory disorders and chronic kidney disease. Ultimately, adults with CHD undergo the pathological progression of their heart disease, in addition to the common cardiovascular risk factors and consequent organ injury. This certainly explains the difficulty and complexity in the management of these patients by nonspecialized medical teams.
Patients admitted to the emergency departments had simple as well as complex ACHD. Primary reasons for presentation to the emergency department differed among these patients but were mainly described as “nonspecific chest pain and respiratory disorders.” Some reasons were more specific like arrhythmias, acute myocardial infarction, PHT, and endocarditis (Fig. 5.2). Pregnancy was often considered as an “illness” in these patients, and in cases of complex CHD, it was managed like a life-threatening condition [4].
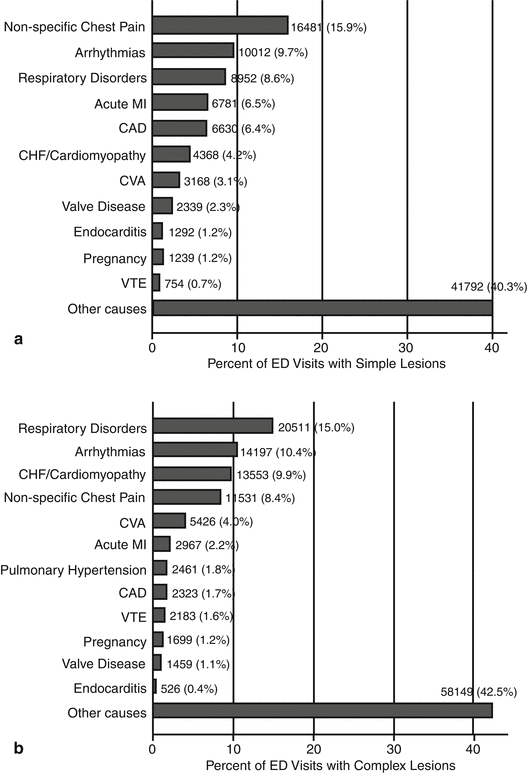
Fig. 5.2
This figure demonstrates the primary reasons for presentation to the ED among patients with (a) simple ACHD and (b) complex ACHD. Abbreviations: ACHD adult congenital heart disease, CAD coronary artery disease, CHF congestive heart failure, CVA cerebrovascular accident, ED emergency department, MI myocardial infarction, VTE venous thromboembolism (Reproduced from Agarwal S et al. [4])
5.2.2 Common Issues
5.2.2.1 Mortality
The reported mortality associated with CHD has decreased during the last 3 years, and survival rates have significantly improved for simple heart defects, as well as for more complex forms, like tetralogy of Fallot and transposition of the great arteries. Nevertheless, the mortality rate for adult patients with Eisenmenger syndrome remained high [5–7].
A recent single-center retrospective study identified the leading causes of death among 7000 adult patients with CHD: chronic heart failure (42%), pneumonia (10%), sudden cardiac death (7%), cancer (6%), hemorrhage (5%), and perioperative mortality [8].
In a literature review, Drenthen demonstrated that almost 11% of women with CHD suffered from cardiac complications during pregnancy. Arrhythmias were identified in 4.5% of the cases. Pregnant women with Eisenmenger syndrome presented frequently with a stroke and died from cardiovascular complications [6, 9].
5.2.2.2 Morbidity
Comorbidities and complications like arrhythmias, heart failure, or endocarditis are common in adult patients with CHD. Pulmonary arterial hypertension is also a serious burden and might progress to Eisenmenger syndrome.
Pregnancy represents a major challenge in the care of female patients with ACHD. The risks of mortality and morbidity, as well as the impact of possible surgeries or interventions, complicate the delicate management of these patients.
Cardiac catheterization and surgery are also increasing; this is mainly due to residual heart defects, the need for valve replacement, and in some cases palliating anomalies when complete repair is not possible.
Holst KA et al. demonstrated that arrhythmias, low cardiac output syndrome, strokes, hemorrhages, ventilation disorders, and kidney injuries may complicate surgery in 15–25% of the patients. Furthermore, perioperative mortality is also estimated to range between 3% and 8% [11].
5.2.2.3 Arrhythmias
Arrhythmias are the most common long-term complication and represent the leading cause of morbidity and death as well as the primary reason for hospital admission. Walsh EP has shown that sudden death is significantly more frequent in ACHD patients than in the general population, with an incidence of 0.09% per year. The prevalence of tachyarrhythmias or bradyarrhythmias is correlated with age and complexity of the heart disease [10].
Sinus node dysfunction and atrioventricular (AV) block are common in the postoperative period. In some cases, AV block will need an implantable permanent pacemaker [12]. Congenital AV block can occur in 3–5% of congenitally corrected transposition of the great arteries, with an increase of 2% each year [10, 12].
Almost 20% of patients with Ebstein’s anomaly develop associated arrhythmias, related to the structural defect itself, like Wolff-Parkinson-White. Surgical repair for Fontan, ASD, tetralogy of Fallot, and Mustard and Senning procedures are frequently complicated with flutter and atrial arrhythmias (tachycardia or bradycardia) [12]. Medical management of supraventricular tachycardia (SVT) is rarely efficient and will require catheter ablation in most cases, although associated with recurrent episodes of SVT [13, 14].
In congenital mitral and aortic valve disease and in single ventricle, atrial fibrillation is frequent (20–30%) [15].
Patients with repaired tetralogy of Fallot are at high risk (2%) of sudden death secondary to SVT, 20 years following surgical repair [16]. These particular patients must undergo meticulous investigations to determine the risk factors that require an implantable defibrillator.
The Heart Rhythm Society, with the collaboration of the American College of Cardiology, the American Heart Association, the European Heart Rhythm Association, and the Canadian Heart Rhythm Society, has suggested recommendations for the management of arrhythmias in patients with ACHD [17].
5.2.2.4 Heart Failure
The pathophysiology of heart failure associated with ACHD is complicated: it is attributed to a number of interrelated mechanisms. The ventricles (especially the right ventricle) suffer from volume overload in an atrial septal defect (ASD) and from volume and pressure overload in a ventricular septal defect (VSD). A single ventricle, in its various forms, might not be capable of responding to long-term metabolic demands. Pulmonary blood flow and lung perfusion are impaired in cases of anomalies in the pulmonary outflow tract. The AV conduction system can be compromised in these patients.
ACHD-associated heart failure is the most common reason for hospitalization in ACHD patients. Hence, this condition should be considered carefully and managed differently from heart failure associated with acquired heart disease.
ACHD patients with cyanotic single ventricle physiology suffer mostly from systolic dysfunction [18]. In these particular patients, mortality and arrhythmias are associated with diastolic dysfunction.
Aggravating factors in ACHD-associated heart failure may be related to many complex parameters: arduous diagnosis, tedious medical and surgical care, and challenging and specialized lifelong follow-up. However, difficulties associated with activities of daily living and with moderate to intense exercise should alarm the specialized medical team. These patients have characteristic lower physical capacity [19].
5.2.2.5 Pulmonary Arterial Hypertension
PAH is associated with ACHD in 4–10% of the cases. Delayed management of left-to-right shunts (VSD, patent ductus arteriosus, truncus arteriosus, aortopulmonary window, and even ASD in some cases) could be responsible for PAH, especially with underlying genetic predisposition to PHT.
Pulmonary endothelial dysfunction and remodeling, worsened by inflammation and infection, will lead to an increase in pulmonary vascular resistance (PVR). With an adequate and maintained right ventricle function, PHT will develop secondary to increased PVR. However, high PVR can lead to right ventricle dilation and dysfunction [20].
Eventually, PVR and PHT become fixed and progress to Eisenmenger syndrome. Multi-organ involvement is usually present, and mortality rate is highly increased when ACHD patients develop Eisenmenger syndrome. Major advances in pulmonary vasodilator drugs may improve survival [21]. CHD with left-to-right shunt are currently being repaired as soon as possible, practically always before the age of 9 months, an approach that has decreased the incidence of Eisenmenger syndrome. However, there are cases of PAH in ACHD, not related to the initial heart defect.
5.2.2.6 Infective Endocarditis
The complexity of surgical repair and the implantation of foreign material or grafts favor the development of infection. Endocarditis in ACHD patients is 20 times more frequent than in the general population [21, 22, 47]. Management of these patients has also improved, decreasing the mortality rate by 10%. However, surgery remains necessary in more than 30% of the cases.
Because of the growing crisis of antibiotic-resistant infections, antibiotic prophylaxis prior to dental procedures is indicated only for ACHD patients with foreign material (i.e., prosthesis) or patients with partially repaired CHD. Moreover, prophylaxis has been limited to 6 months following surgical intervention. Antibiotic prophylaxis is not recommended anymore before pulmonary or respiratory track procedures, neither for digestive nor genital and urinary track interventions. Tattoos and piercings should be avoided in ACHD patients [23].
5.2.2.7 Pregnancy
Fetal mortality and premature birth represent almost 4% of all births from ACHD, making it four times higher than in the general population. Maternal and fetal risks are both increased in pregnant women with ACHD. Eleven percent of these women present cardiac complications, particularly in those with PAH and in some complex situations like outflow tract stenosis (pulmonary and aortic), pulmonary atresia, left AV valve stenosis, residual coarctation of the aorta, bicuspid aortic valve with dilated aortic root, and Marfan and Turner syndromes [9, 24]. In women with an arterial oxygen saturation of less than 85%, only few pregnancies are successful [25]. The most common maternal risks are heart failure (4.8%) and arrhythmias (4.5%). Women with ACHD and PAH can present with acute cardiac decompensation during pregnancy, especially around 25 weeks of gestation. This risk is increased with inflow obstructive lesions of the systemic ventricle. Preeclampsia, eclampsia, and thromboembolic events are also more frequent in women with ACHD.
ZAHARA study identified risk factors during pregnancy in women with ACHD: cyanotic heart disease, mechanical valve replacement, and systemic or pulmonary AV valve regurgitation (Table 5.1) [9].
Table 5.1
Scores predicting maternal cardiovascular complications during pregnancy (Reproduced from Ntiloudi [6])
ZAHARA | |
• Prior arrhythmia | 1.50 |
• NYHA functional class III/IV | 0.75 |
• Left heart obstruction (peak LVOT gradient >50 mmHg or aortic valve area <1.0 cm2) | 2.50 |
• Mechanical valve prosthesis | 4.25 |
• Systemic AV valve regurgitation (moderate/severe) | 0.75 |
• Pulmonary AV valve regurgitation (moderate/severe) | 0.75 |
• Cardiac medication before pregnancy | 1.50 |
• Cyanotic heart disease (corrected and uncorrected) | 1.00 |
5.2.2.8 Hemostatic Disorders
Thromboembolic events are 10–100 times more frequent in patients with ACHD. A study on 23,150 patients with ACHD demonstrated that 2% suffered one or more strokes (0.05% event per year per patient). Patients with cyanotic heart disease and reduced systemic arterial oxygen saturation are the most exposed to cerebral thrombotic events [26, 48].
There are currently no absolute guidelines or recommendations for anticoagulation strategies in these patients. Individualized approach and management should be considered to prevent thromboembolic events [27].
5.2.3 Anesthesia and Postoperative Care
The management of ACHD patients following surgical interventions is complex and challenging. Specialized teams of anesthesiologists and intensivists are imperative. A scrupulous understanding of the patient’s structural heart defect and his functional status following the surgical repair or the cardiac catheterization is one of the most important aspects in the management of these particular patients.
Careful acknowledgment of the cardiac output, the systemic vascular resistances, and the heart-lung interaction is fundamental for the majority of congenital heart diseases, at all times.
Vascular access might be problematic due to the previous numerous surgical procedures in many of these patients. Moreover, reoperation is associated with increased mortality [28]. Management strategies should be well established in order to prevent and avoid complications and to provide patients with rapid and correct medical care and treatment.
5.2.4 Physiology
Patients with decreased pulmonary blood flow secondary to right ventricular outflow tract and pulmonary valve obstruction (i.e., patients with tetralogy of Fallot) are dependent upon preload conditions. Thus, maintaining relatively high blood pressure and cardiac output and avoiding increased PVR and oxygen consumption are very important during the perioperative period.
An opposite situation is that of a patient with increased pulmonary blood flow (i.e., patients with VSD); this patient has a compromised systemic perfusion that might lead to left heart failure.
In these two situations, pulmonary-to-systemic blood flow ratio should be maintained constant at all time, by optimizing the cardiac output, volemia, and the pulmonary and systemic vascular resistances.
5.2.4.1 Preoperative Assessment
Thorough and meticulous understanding of the initial cardiac defect and its progression is a must for the anesthesiologist. He should also well identify previous surgeries and interventions. Complete clinical and biological evaluation should be performed prior to cardiac, as well as noncardiac surgery. Identifying comorbidities, in particular performing pulmonary function test to detect respiratory problems or stress test in some cases, has an impact on the management of these patients. ACDH patients have limited physical capacities, which could compromise the weaning from mechanical ventilation. Ultrasound evaluation of vascular access should be performed to rule out thrombosis due to frequent catheterizations and surgeries and to decide upon the sites for central and arterial lines.
5.2.4.2 Anesthetic Management
Sedative premedication is necessary. Nevertheless, it should be administered with caution in case of hypoxemia. Selection of the sedative agent should be individualized.
Inhaled anesthetic agents used for induction may have a delayed result in case of decreased pulmonary blood flow. Increasing the agent’s concentration should be avoided because of its toxicity. Patience and more time are needed for these patients with congenital heart disease.
In addition, the majority of anesthetic agents produces systemic vasodilation and has negative inotropic effect on the heart, compromising the delicate balance of pulmonary-to-systemic blood flow ratio in case of intracardiac shunts.
Intravenous induction should also be performed with caution and should be associated with volume and fluid expansion.
Related posts:
 Patients with Cerebral Diseases
The Patient with Septic Shock
Anaesthesia and Rare Metabolic Disorders
Patients with Cerebral Diseases
The Patient with Septic Shock
Anaesthesia and Rare Metabolic Disorders
 Obstructive Sleep Apnea (OSA) and Anesthesia
Obstructive Sleep Apnea (OSA) and Anesthesia
 The Severely Obese Patient
The Severely Obese Patient
 What Every Anaesthetist Needs to Know About Respiratory and Cardiovascular Dynamics in Patients with Obesity or Intra-abdominal Hypertension
What Every Anaesthetist Needs to Know About Respiratory and Cardiovascular Dynamics in Patients with Obesity or Intra-abdominal Hypertension
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


