Opioid
Year synthesized
Chemist and/or companya
Oxymorphone (Numorphone)
1914
Oxycodone
1916
Freud and Speyer, University of Frankfurt, 1917
Hydrocodone
1920
Mannich and Lowenheim
Hydromorphone (Dilaudid)
1924
Launched by Knoll, 1926+
Meperidine (pethidine-Demerol)
1932
Eisleb, launched by IG Farber 1938–1939+
Methadone
1937
Launched by IG Farber, 1939+
After the war, the Allies confiscated German patents, trademarks and research records. Some research records were slowly released over the next two decades, slowing the post-war introduction of these opioids into the UK, and North and South America. In contrast, the availability, use, and experience of clinicians in Continental Europe with the German opioids before, during and after World War II, allowed physicians in these countries to become familiar with intravenous opioids and opioid-like analgesics as analgesic supplements. This accelerated European acceptance of the Janssen opioids introduced in the late 1950s–1980s. First came dextromoramide (Palfrum), followed by phenoperidine and then fentanyl, alfentanil and sufentanil, some mixed with new intravenous hypnotics, tranquilizers, sedatives, and amnestics as early forms of balanced anesthesia. The successes of German companies with the new opioids, and the popularity of ultra short acting barbiturates stimulated many European pharmaceutical companies to pursue the development of similar compounds from the late 1940s through to the 1960s.
During this period, the situation in the US and the UK differed significantly from that in Europe. The German opioids synthesized between 1914 and 1937 were either not available or had limited impact on anesthesia practice (perhaps partly due to surgeons’ resistance to the use of intra-operative opioids as mentioned previously) in these countries. In addition, most companies in the US and UK anesthesia related business (British Oxygen Company, Ohio Medical, von Foregger, Heidbrink, McKesson and others) sold anesthetic machines which delivered anesthetic gases contained in tanks (also sold by the companies) or volatile liquids delivered from vaporizers on their machines. Inhalation anesthesia was a US and UK invention, and to an extent is still a more important part of anesthetic practice in the US and UK than intravenous anesthesia, the reverse being true in Europe (see section on total intravenous anesthesia). Finally, the capital cost of the machines required to deliver an inhaled anesthetic exceeded that required to deliver an intravenous anesthetic, a significant factor for Europe, recovering from an economically disastrous war. Thus in the first two decades after World War II, European but not North American companies vigorously pursued development of new intravenous opioid and non-opioid anesthetic compounds.
Neurolept Analgesia’Anesthesia
In the late 1940s and early 1950s, European investigators re-examined inhalation anesthesia [4,29–33], believing they might create a better state of general anesthesia with less organ system depression by substituting or adding intravenous compounds. The success of the barbiturates in the 1930s and early 1940s stimulated several European companies to look at other “intravenous anesthetics” as potential alternatives to inhalation agents. These approaches sought a “better anesthetic state,” one that provided analgesia (and thus was more effective at blocking the “stresses of surgery”), anxiolysis, sedation and hopefully amnesia without disturbing (or minimally altering) cardiovascular, respiratory, and other vital organ functions [4,29,30].
In 1954 in France, Laborit and Huygenard introduced the idea of artificial hibernation [29]. They attempted to inhibit cellular, autonomic, and endocrine mechanisms that are normally activated in response to stress. They developed a “lytic cocktail” containing an analgesic (meperidine), two tranquilizers (chlorpromazine and promethazine), and atropine [29,30]. The resultant “artificial hibernation” (also called ganglioplegia or neuroplegia) often produced circulatory depression and delayed awakening, and did not achieve widespread popularity [4]. Another approach called “ataralgesia” nominally provided tranquility and freedom from pain by combining an analgesic (meperidine), a tranquilizer (mepazine), and an analeptic (aminophenazole). A related cocktail called “narco-ataralgesia” used diazepam, phenoperidine, and droperidol. Neither was widely accepted for reasons similar to those described above [4].
Janssen synthesized haloperidol, the first member of a new series of tranquilizer compounds called butyrophenones in the late 1950s. It induced a syndrome called “neurolepsis” [4,32] consisting of inhibition of psychic, vegetative, and motor functions (patients became cataleptic) and suppression of apomorphine-induced vomiting. In 1959, de Castro and Mundeleer combined haloperidol with the new narcotic analgesic, phenoperidine in the first demonstration of neurolept analgesia (NLA)’a detached, pain-free state without marked circulatory depression [33]. In the 1960s, NLA with haloperidol and phenoperidine achieved significant popularity in Europe as an alternative to anesthesia using potent inhaled agents [4,31,34]. Janssen and colleagues then produced even more potent drugs, the butyrophenone droperidol, and the opioid fentanyl [4]. In 1963, de Castro and Mundeleer found the fentanyl–droperidol combination superior to haloperidol and phenoperidine for NLA, with more rapid onset of analgesia, less respiratory depression and fewer extrapyramidal side-effects [4,31,34]. Use of NLA with fentanyl and droperidol became widespread throughout Europe in the 1970s and early 1980s. When N2O was added to the mixture in the early 1960s (to improve analgesia and amnesia), the technique was called neurolept anesthesia (NLAN).
Besides droperidol and haloperidol, a phenothiazine (usually chlorpromazine and promethazine) or sometimes diazepam were used with fentanyl for neurolept analgesia in Europe during the 1960s–1980s. Some historians believe the numerous variations in NLA techniques developed during this interval deterred further popularization of the technique. Reasons cited include confusion about the choice, timing, and dosage of drugs to be administered, and the unclear indications for and contraindications to NLA [4]. It is also possible that NLA and NLAN are inferior approaches to anesthesia. That might explain the numerous variants developed in a search for something better. As a result, many European clinicians lost confidence in the efficacy of NLA and NLAN. In the US, neither NLA nor NLAN became popular because of the aforementioned concerns, the lack of experience, and the unavailability of many of the compounds [4,10].
Opioid Antagonists and Agonist-Antagonists
Pohl synthesized the first opioid antagonist, N-allylnorcodeine in 1914 while attempting to improve the analgesic properties of codeine [4,10]. The discovery that this compound mildly antagonized the respiratory depression produced by morphine went unnoticed for 26 years, until McCawley and co-workers (in 1940) in search of a strong analgesic with “built in” antagonistic action, attempted to prepare N-allylnormorphine (nalorphine) [4,10]. Weyland and Erickson successfully synthesized nalorphine in 1942, and then found that the drug blocked all actions of morphine [4,10]. In 1950, Schnider and Hellerback, found that nalorphine possessed strong analgesic properties in humans [4,10,35], but the doses needed to provide analgesia also yielded severe psychotomimetic effects, rendering it clinically unsuitable as an analgesic.
In 1960, naloxone was synthesized and found to be a more potent and less toxic opioid antagonist than nalorphine [4,10,36]. Naloxone produced no dysphoria at any dose but had a short duration of action [35]. By the 1970s, it had largely replaced nalorphine in the peri-operative period [4,10]. In the 1980s, a longer lasting pure opioid antagonist called nalmefene was introduced for clinicians needing prolonged opioid antagonistic activity [10].
The discovery of nalorphine stimulated a search for other agonist-antagonist drugs in the 1950s. Most chemists focused on replacing the methyl (CH3) group attached to the N of the piperidine structure in morphine (Fig. 48.1) or oxymorphone. Pentazocine (also known as Fortral or Talwin) was the first successful agonist-antagonist produced [4,10,37]. Synthesized by the Sterling-Winthrop Research Institute of the Sterling Drug Company of Rensselaer, New York in 1958, it was approved by the FDA in 1967 and released in mid-1967 in the US, England, Mexico, and Argentina. The hope was that pentazocine (which acts principally at k rather than at µ receptors) would provide analgesia with less respiratory depression than morphine, and less liability for abuse. Although the potential for abuse is less and a ceiling for respiratory depression does occur at doses of 30–70 mg, the compound is only one-half to one-fourth as potent as morphine, resulting in maximal analgesia at a dose similar to that causing respiratory depression. It can also cause dysphoric side effects similar to those of nalorphine. None of the other agonist-antagonists found clinical or commercial success.
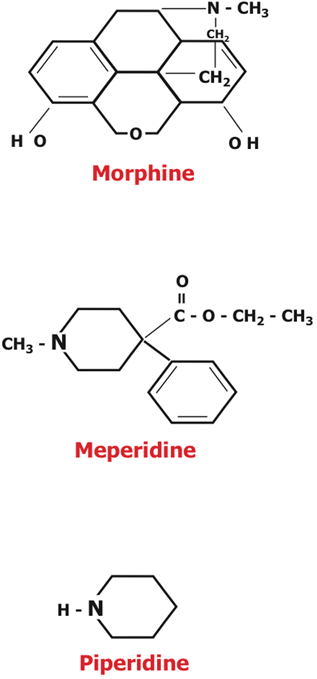
Fig. 48.1
Morphine and meperidine shared characteristics that made them analgesics and provided clues in the search for a better opioid analgesic
Re-introduction of High-Dose Opioid Anesthesia
Morbidity and mortality, likely from inadequate ventilation, caused the abandonment of twilight sleep by 1915. By the 1960s, assisting and controlling ventilation had become commonplace, enabling the re-introduction of high dose opioid administration. In the early 1960s, the cardiac anesthesia group at the Massachusetts General Hospital (MGH) in Boston, and de Castro in Brussels independently experimented with high-dose “opioid anesthesia” [38–41]. The Boston group used morphine and de Castro used fentanyl. Both used opioids to produce anesthesia without compromising cardiovascular stability. Their patient populations differed however: the sickly Boston patients had surgery for end stage cardiac valvular disease while De Castro’s had routine surgical procedures such as cholecystectomy or bowel resection.
The cardiac anesthesia group at MGH contended with end stage rheumatic aortic and/or mitral valvular heart disease, low cardiac indices, and pulmonary dysfunction [38,39]. The then standard induction of anesthesia, carefully performed with thiopental and succinylcholine, followed by N2O, halothane and curare, frequently produced hypotension and arrhythmias, and cardiac arrest was not uncommon. Death during or soon after surgery was distressingly frequent. All patients required mechanical ventilation in the Respiratory Care Unit and most received morphine to enable them to tolerate their tracheal tubes. The anesthesiologists caring for these patients made a surprising observation: tens of milligrams of morphine were usually required for tracheal tube tolerance in the post operative period, but these huge doses had minimal circulatory effects, and often resulted in unconsciousness [38,39].
From this observation, Myron Laver, one of the cardiac team, reasoned that morphine might substitute for barbiturates and inhalation anesthetics for induction of anesthesia, and the team moved from small to large doses of morphine, first with additional inhalation anesthetics and then with only oxygen plus a neuromuscular blocking agent and scopolamine. Few of the patients anesthetized with the new technique reported awareness or pain. The high-dose morphine technique changed induction of anesthesia from an anxiety filled period to a calm controlled one that also set the stage for an orderly transition to postoperative mechanical ventilation.
After using the technique in 1,000 patients, the MGH cardiac anesthesia team formally studied 15 patients (7 with aortic-valve disease and 8 without major heart or lung disease) [39]. Published in December of 1969, the report confirmed the benign nature of a large dose of morphine in severely compromised patients. The 15 patients breathed 100% oxygen while 1 mg/kg of morphine was slowly infused [39]. The anesthetist had to shout at the patient to prompt continued breathing. Hemodynamics, measured frequently, remained stable, and additional morphine, sometimes up to 3.0 mg/kg was given. As the patients became unresponsive, they were paralyzed and the trachea intubated. Results of the study were “startling and pleasing” [38,39]. The control patients (no heart or lung disease) had no consistent hemodynamic response; those with aortic valve disease experienced increased cardiac output and decreased systemic vascular resistance. The report changed the practice of anesthesia for patients with severe cardiac disease. Within months, clinicians throughout the US experimented with high-dose morphine-oxygen anesthesia in patients having cardiac surgery, and similar patients having major non-cardiac procedures, and reported positive results. Lowenstein’s classic article was rejected for presentation at the annual meeting of the American Society of Anesthesiologists.
Meanwhile in Brussels in the early 1960s, de Castro looked for ways to provide anesthesia that minimally altered cardiovascular dynamics and also blocked the “stress hormonal” responses to major surgery [40,41]. He was not a cardiac anesthesiologist but did provide anesthesia for patients undergoing most kinds of surgery. He used large doses of the newly-introduced fentanyl (up to 50 μg/kg) plus oxygen to produce what he called analgesic anesthesia [40,41]. He gave analgesic anesthesia to patients having cholecystectomy, gastric resection, bowel surgery, and similar operations, finding minimal cardiovascular and stress hormonal changes during and after surgery. He suggested that analgesic anesthesia had advantages beyond minimal cardiovascular and stress hormonal changes; simplicity (no need for hypnotics); lack of side effects (no histamine release which can occur after morphine) and a high therapeutic index (safety margin 400 vs. 70 for morphine). His patients did not report awareness but their lungs often required mechanical ventilation for up to 3 hours after surgery, before tracheal extubation could be accomplished. He reported his results at the World Congress of Anesthesiology in Mexico City in 1976 [40]. He was unable to publish his results in a major anesthesia journal but did publish them in a regional European journal [41]. Because his work remained unknown to most anesthesiologists, it had little impact on the world anesthesia community.
Between December 1969 and 1975, numerous reports extolled the virtues of morphine-oxygen anesthesia in severely ill patients [42–47]. Stanley studied the effects of high (1–3 mg/kg) and ultra-high (8–11 mg/kg) doses of morphine in patients undergoing cardiac surgery, finding advantages and disadvantages [43,44]. The ultra-high doses decreased the need for non-opioid pharmaceutical supplementation and ensured that patients were “unaware”, but produced increased veno-vasodialation, requiring markedly greater crystalloid and or/colloid fluid replacement to maintain stable intra and postoperative hemodynamics and adequate urinary output. The greater infusion of fluids increased tissue edema. High dose morphine anesthesia became particularly popular in sick patients having valvular heart surgery [4,10,42–49]. However, nothing’s perfect! Problems with incomplete amnesia, histamine release, prolonged postoperative respiratory depression, increased blood volume requirements secondary to marked venodilation, and hypotension and hypertension limited the popularity of morphine as a sole anesthetic [4,10,42–49].
The problems with high-dose morphine prompted studies of high-dose fentanyl anesthesia in animals [50–53], and then in patients having first valvular and later coronary artery surgery [54–56]. Clinical success in the late 1970s and early 1980s dramatically increased fentanyl usage. Sales in the US increased 10-fold during the first year the drug was off-patent (1981) [34]. Why? Before the reports of high-dose fentanyl anesthesia, fentanyl was infrequently used in a dose exceeding 50 μg for an entire operation. However, after the reports, fentanyl doses used in cardiac operations increased to 50–100 μg/kg [34,42]. High-dose fentanyl anesthesia rapidly replaced high-dose morphine for both cardiac patients having valvular heart surgery and patients undergoing the new coronary artery bypass operations in the 1980s [10,42]. Fentanyl’s advantages over morphine were its greater potency and ease of use (it could be safely administered rapidly in a minute or less), its shorter onset and duration of action, and an absence of histamine release and venodilation. As a result, induction of anesthesia was faster. There was less hypo and hypertension, blood and crystalloid volume requirements were not increased, and tracheal extubation and post-operative recovery occurred sooner [10,42].
The marked increase in fentanyl usage throughout the world in the early 1980s spurred the Janssen Company to develop sufentanil and alfentanil [4,10,34,42,57–61]. Glaxo experimented with new opioids, resulting in remifentanil [62], and Anaquest of the British Oxygen Company developed its own series of opioids that are on the brink of being introduced for wild life immobilization [63]. Alza and Anesta (young drug delivery companies in the mid 1980s) began experiments with fentanyl in transdermal patches and oral mucosal lozenges [64–68].
Paul Janssen and the Fentanyl Family of Analgesics
Janssen (“Dr. Paul” to friends and colleagues) was born (1926), raised, and educated in Belgium. After graduating from medical school, he gained a PhD while working with Nobel Prize winner Corneille Heymans at the University of Ghent [34]. His father, Conrad Janssen, much influenced Dr. Paul. His father started his professional life as a family doctor, later selling pharmaceutical products [34], mostly tonics, stimulants, vitamin preparations and organic extracts. During college, Dr. Paul, perhaps because of exposure to his father’s business, saw the importance of chemistry to medicine, what he called “medicinal chemistry.” He realized that selling only generic medicines limited the future of the company, and this heightened his interest in new drugs. He also understood that chemical structure determined action. In 1953, after visiting several US and European pharmaceutical companies and pharmacologists, Dr. Paul started Janssen Pharmaceutica. An early interest was the pain produced by muscle spasms. In 1955, he introduced the fifth of his newly synthesized compounds, ambucetamide (Neomeritine), a uterine antispasmodic still marketed for the relief of menstrual pain.
In 1953, morphine (Fig. 48.1) was the standard analgesic. Dr. Paul knew about the synthetic alternative, the less potent meperidine (see above). Both meperidine and morphine contain a piperidine ring (Fig. 48.1), and Dr. Paul conjectured that this was important to the production of analgesia. Working with meperidine as the lead molecule (because it was less complex and easier to manipulate than morphine), they began a search for molecules that were more powerful and specific analgesics, molecules with fewer unwanted side effects [34].
In 1956, the Janssen team thought that meperidine was one-tenth as potent as morphine because it poorly penetrated the blood-brain barrier.1 To overcome this limitation, they believed a more fat soluble derivative was needed, leading them to replaced the methyl (CH3) group attached to the N at the extreme left of the meperidine molecule (Fig. 48.1) with a benzene ring. They then added a C=O in combination with two CH2 groups between the left benzene and piperidine rings of the first new compound. The resulting compound R951 (Fig. 48.2), showed a greater analgesic potency. Addition of another CH2 group between the benzene and piperidine rings in R951 produced R1187 (Fig. 48.2), a less analgesic compound than R951. In 1957, Janssen chemists changed the C=O group attached to the left benzene ring in R951 to a C–OH group, producing R1406 (phenoperidine; Fig. 48.2). At that time phenoperidine was the most potent opioid in the world, having 20 times the potency of morphine [34]. It was introduced into Europe in 1964, but not the US, as a potent, fast acting, short lasting analgesic for anesthetic use. It is still used in Europe.
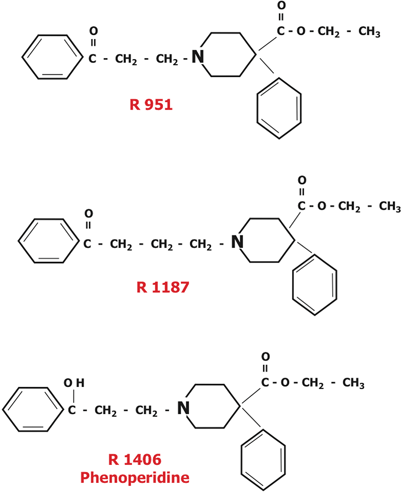
Fig. 48.2
Various experimental steps led to a new opioid, and ultimately to the useful series shown in Fig. 48.3
In 1960, the Janssen team modified the phenoperidine molecule to produce fentanyl (Fig. 48.3). Fentanyl was approved for use in Europe 3 years later [34,57]. Subsequently, the Janssen team created sufentanil (synthesized in 1974, introduced into Europe in 1979 and the US in 1985), alfentanil (synthesized in 1976, introduced into Europe in 1983 and the US in 1987) and carfentanil (synthesized in 1974 introduced into veterinary [wild life immobilization] medicine in 1986; Fig. 48.3; Table 48.2) [57–61].
Table 48.2
Potency and safety comparisons of morphine, meperidine and some Janssen analgesic compounds. (From Janssen Pharmaceutica and de Castro et al. [60], with permission)
Compound | Tail withdrawal reflex ED50 (mg/kg) | Potency ratioa | LD50 (mg/kg) | Therapeutic index |
|---|---|---|---|---|
Meperidine | 6.15 | 1.00 | 29.0 | 4.72 |
Morphine | 3.15 | 1.95 | 223 | 71.0 |
Phenoperidine | 0.12 | 51.3 | 4.69 | 39.1 |
Alfentanil | 0.044 | 140 | 47.5 | 1,080 |
Fentanyl | 0.011 | 559 | 3.05 | 277 |
Sufentanil | 0.00067 | 9,180 | 17.9 | 26,700 |
Carfentanil | 0.00037 | 16,200 | 3.13 | 8,460 |
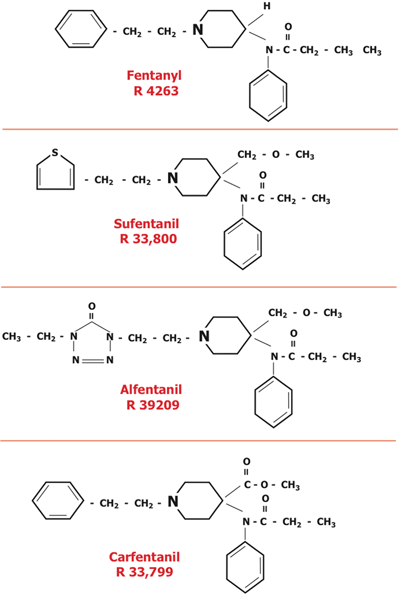
Fig. 48.3
Clinically useful opioids that resulted from the steps suggested in the previous figure. The first three were used in humans, the last, carfentanil, was used to produce immobilization in animals
Fentanyl’s Approval in the US
The international healthcare conglomerate, Johnson & Johnson (J&J), purchased Janssen Pharmaceutica in July 1961 [34], providing Janssen with financial security and access to individuals within the J&J family, including Robert McNeill, the founder of the Philadelphia Company, McNeill Laboratories, purchased by J&J in 1959. In the mid 1960s, Janssen successfully launched fentanyl and droperidol as independent drugs in Europe [32–34,57]. Janssen could not however, get Food and Drug Administration (FDA) approval for fentanyl in the US [34]. Robert Dripps, Professor of Anesthesiology at the University of Pennsylvania, felt that fentanyl was too potent, and caused muscle rigidity. These effects, he thought, increased the need for tracheal intubation and would lead to abuse problems. McNeill knew Dripps and introduced Dripps to Janssen, beginning a dialogue that led to a compromise. Fentanyl would be submitted for approval in combination with droperidol. When approved by the FDA in 1968, fentanyl became available combined with droperidol in a ratio of 50:1 droperidol to fentanyl. The combination was called Innovar in the US and Thalamonal in other countries.
Why with droperidol, and why a 50:1 ratio? Janssen had consulted with Belgian anesthesiologist, George de Castro [34]. De Castro had tested Janssen’s intravenous (IV) analgesics, hypnotics, and sedatives in patients’after animal testing in the Janssen laboratories. He used fentanyl in combination with droperidol in a neurolept-anesthesia technique popular in some Western European countries in the early to mid 1960s. Reviewing his clinical practice, he found that he used fentanyl and droperidol in approximately a 50:1 ratio, the ratio then suggested by Janssen to Dripps. Both knew that the recreational use of droperidol produced a “bad high” and believed that mixing droperidol and fentanyl would minimize any abuse potential. The FDA agreed and Innovar was approved for use in the US in early 1968 (personal communication, P. Janssen and T. Stanley). Four years later, fentanyl became available alone but for the next six years only as a 1 ml vial containing 50 µg [34].
Opioids as Supplements to Induction of General Anesthesia
Todays anesthetists often use opioids to enhance the induction of general anesthesia [10,42,62,69,70]. They use doses of opioids during induction that exceed those used for entire surgical cases in the 1950s, 1960s and 1970s, thereby providing analgesia and decreasing tachycardia and hypertension during induction (including rapid sequence induction) and maintenance of anesthesia. Such administration also reduces the requirements for hypnotics during anesthetic induction and maintenance as well. Such benefits explain the present popularity of opioid use [10,42].
The transition from infrequent small doses of opioids during the 1960s and 1970s to the more frequent and larger doses used today took place largely during the 1980s [10,42]. In November of 1979, Ted Stanley, having introduced high-dose fentanyl and oxygen anesthesia in the US [54–56], was invited to join Simon de Lange at the University of Leiden to evaluate the then new opioids, sufentanil and alfentanil. They first visited de Castro in Belgium since he had great experience with these and most other opioids. They discussed the properties and advantages of alfentanil and sufentanil with de Castro and watched him administer both agents during various surgical procedures in order to gain insight into his experience with them.
De Castro was most impressed with alfentanil because of its rapid onset of action (60–90 s) and short duration (3–5 min) after a bolus injection. In December 1979, he suggested that alfentanil should be effective as a sole anesthetic induction agent, as an analgesic component of nitrous oxide–opioid “balanced” anesthesia, and possibly as a sole opioid anesthetic for cardiac surgery. He was also impressed with the potency and ability of relatively high doses of sufentanil to block hemodynamic and hormonal responses to “surgical stress.” He thought it would be useful as a sole or principal opioid anesthetic for cardiac and major vascular surgery. De Lange, Stanley, Stanski and others began a series of studies with alfentanil and sufentanil in January 1980 that spread to other centers in the US and Europe, changing the way clinicians viewed and used opioids [71–80]. It became clear that both alfentanil and sufentanil could be administered (like fentanyl) in doses that resulted in unconsciousness in less than five minutes (<2 min with alfentanil) without appreciable change in heart rate and arterial blood pressure.
Publication of hundreds of papers in the 1970s studying large doses of morphine as an “anesthetic”, and then similar studies with fentanyl in the late 1970s and early 1980s, began a new anesthetic paradigm. This continued with studies evaluating alfentanil and sufentanil as induction agents, as sole opioid anesthetics for cardiac surgical and other procedures in the 1980s. With regulatory approval and introduction of alfentanil and sufentanil into clinical practice in the mid-1980s, clinicians (especially those in the UK and US) became more comfortable with larger doses of the potent opioids. While no opioid became popular as a “sole induction agent” (other than in high-dose opioid–oxygen anesthesia for cardiac or major vascular surgery), a large dose use of these compounds to provide analgesia before and during induction/intubation and during the maintenance of anesthesia, did catch on.
Sufentanil
The remarkable increase of sales in fentanyl as it went “off patent” stimulated sales of generic versions of the drug in the early 1980s. Concurrently, the Janssen research and development team had another newer opioid, sufentanil (Fig. 48.3), that they felt was superior to fentanyl and had a patent life that extended until the mid 1990s [34]. They believed sufentanil would be a better opioid for cardiac and major vascular surgery than fentanyl because early studies indicated that sufentanil provided more cardiovascular stability and greater suppression of hormonal stress responses than fentanyl [10,42,71]. In addition, sufentanil was significantly more potent (which at the time was considered the explanation for its hemodynamic and stress hormonal superiority), had a markedly higher therapeutic index (which they reasoned would make it a safer drug for clinicians), had a similar or possibly faster onset time and shorter duration of action than fentanyl, and did not increase plasma histamine or cause venodilation [34,42,71,72].
While subsequent studies generally confirmed the advantages of sufentanil over fentanyl [81,82], its approval by American and European regulatory authorities (in 1982 in Europe and 1985 in the US) and introduction into clinical practice, did not have a major impact on clinicians. Sufentanil was usually priced significantly higher than fentanyl, impeding its acceptance. In addition, the introduction of the beta blocking drugs at about the same time neutralized sufentanil’s hemodynamic and stress hormonal advantages over fentanyl. While sufentanil did and still does find a place in many cardiac anesthesiologists’ practices, it was never a major commercial success. Sufentanil and alfentanil’s poor commercial performances in the late 1980s and early 1990s convinced the Janssen company that further research in opioids for anesthesiology was not worthwhile. The similarly poor commercial performances of etomidate and droperidol caused the company to leave the anesthesiology field entirely in the mid 1990s.
Alfentanil
Janssen synthesized alfentanil in 1976 and introduced it in Europe in 1983, and in the US in 1987 (Fig. 48.3). Alfentanil offered a rapid onset (60–90 s), short duration (5–10 min when used as a bolus) and a high therapeutic index (1080), (Table 48.2) [34,42,73–79]. These features caused the Janssen Company to envision alfentanil as the perfect opioid for short surgical and outpatient procedures and the increasingly popular total intravenous delivery of anesthesia [34,42]. When alfentanil was introduced in the US in 1987, the possibility of a “blockbuster” was considered likely. It didn’t work out that way.
Although a large bolus dose of alfentanil produced unconsciousness in a minute or so, it also produced muscle rigidity in 70–80% of patients [74]. As a result, alfentanil did not become popular as an induction agent. Alfentanil’s quick onset and short duration has been valuable in single or few bolus doses as a sole analgesic agent or in combination with inhaled or intravenous hypnotics for balanced anesthesia. However, when used for 45–60 min or longer, either as a continuous infusion or as many boluses, alfentanil accumulates in tissues and its duration becomes prolonged [79,80,83]. This made alfentanil less desirable for TIVA (see below) especially relative to remifentanil which was introduced into clinical practice in 1996 [84–86]. Alfentanil has a niche today as a rapid onset/offset analgesic IV bolus prior to short painful procedures [10], but even in that application, remifentanil often supplants alfentanil.
Total Intravenous Anesthesia (TIVA) and Opioids
By definition, TIVA is used for both anesthetic induction and maintainance with intravenous drugs alone [87]. Therefore TIVA excludes the use of nitrous oxide or volatile anesthetics. While the first intravenous anesthetic, chloral hydrate, introduced by Pierre-Cyprien Oré in Paris in 1872, could produce complete anesthesia, mortality associated with its use dampened enthusiasm for its application and for the development of other intravenous anesthetics until the twentieth century [87]. The first intravenous non-opioids developed for anesthesia in the early 1900s (hedonal, a urethane derivative introduced in 1909) and the early barbituates (in the beginning of the 1920s) were slow in onset, had a long duration of action (or both) and were not successful in clinical practice [87]. The 1930s introduction of the ultra-short acting barbituates changed anesthetic induction in adults.
The re-introduction of opioids into operating rooms, especially in the US and the UK, the increasing popularity of “balanced anesthesia” worldwide, and the introduction of neurolept analgesia (anesthesia) in Europe in the 1950s, set the stage for TIVA. The first TIVA advocates (they did not call it TIVA) introduced “artificial hibernation,” “ataralgesia,” “narco-ataralgesia” and neurolept analgesia. These approaches used an opioid (usually meperidine, phenoperidine, or fentanyl) plus non-opioid intravenous hypnotics (tranquilizers, benzodiazepines and other similar compounds). The theory was to independently regulate each anesthetic component (unconsciousness, amnesia, the sympathetic nervous system, and muscle relaxation) with intravenous agents that targeted the specific component. But the cumulative effects of the intravenous agents of the times and inadequate methods of administration (intermittent-bolus administration) resulted in delayed awakening and often circulatory depression. Accordingly, interest in TIVA waned until the 1980s when the introduction of computerized pharmacokinetic-driven infusion devices combined with better drugs stimulated new research [87].
In 1981, Helmut Schwilden showed that a computer-controlled infusion pump guided by the published pharmacokinetics of the drug could produce target plasma anesthetic (and analgesic) concentrations in specific populations [88]. This began the application of target-controlled infusion (TCI) which is also sometimes called computer-assisted continuous infusion (CACI). Jacobs, Reves and Glass coined the latter terminology in 1991 [89]. With this technique, the anesthesiologist chooses a plasma concentration of both a hypnotic and an analgesic, usually an opioid. The technique accounts for the patient’s characteristics and the degree of anticipated surgical stimulation. The computer-driven infusion pump then adjusts the drug infusion rate to obtain the targeted plasma concentration as predicted by pharmacokinetic information known for a similar population of patients.
In 1992, Shafer and Stanski showed that computer simulation programs of continuous infusions of intravenous anesthetics (including opioids) increased the understanding of these drugs’ clinical pharmacokinetics [90]. It became clear that the terminal half-life of an intravenous anesthetic does not predict the rate of decrease of its plasma concentration after discontinuation of administration. In 1991, Shafer and Varvel found that the duration of drug infusion influenced the immediate rate of decline of fentanyl, alfentanil and sufentanil effect-site concentrations during recovery [91]. In 1992, Hughes and colleagues introduced the concept of “context-sensitive half-times” (the time it takes for the plasma concentration of an opioid to decrease by 50% after its infusion is discontinued). The concept was also confirmed with intravenous hypnotics [92]. In 1994, Young and Shafer proposed the desirable pharmacokinetic properties that an intravenous opioid should posses for rapid onset and recovery [93].
Some advocates of TIVA believe that ultimate acceptance of the technique depends on closed-loop administration [87]. The development of a feedback control system for maintaining neuromuscular blockade using neuromuscular blocking agents (pancuronium) was first described in 1980 and was considered a breakthrough [94]. The absence of an adequate feedback signal for the development of closed-loop systems for the control of other components of the anesthetic state, hypnotic depth, and especially a measure of analgesia, have impeded the development of closed-loop TIVA systems [87,95]. TIVA has become increasingly popular in Europe in the last decade despite less than ideal closed-loop systems. This has resulted from the introduction of short onset, short duration intravenous agents (particularly propofol and remifentanil), and the quantification of their pharmacokinetics, the invention of “brain monitoring devices”, and the development of “smart” infusion pumps.
Related posts:
 History Reflected in the Evolving Approaches to Anesthesia for a Patient Undergoing Cholecystectomy
A History of Intravenous Anesthesia
History Reflected in the Evolving Approaches to Anesthesia for a Patient Undergoing Cholecystectomy
A History of Intravenous Anesthesia
 Major Anesthesia-Related Events in the 2000s and Beyond
Major Anesthesia-Related Events in the 2000s and Beyond
 1846–1860: Following the Discovery of Anesthesia
1846–1860: Following the Discovery of Anesthesia
 A History of Research in Anesthesia
A History of Research in Anesthesia
 The Evolution of Premedication
The Evolution of Premedication
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


