Surfactant: Introduction
Pulmonary surfactant lines the inner layer of the lung and serves to lower surface tension at the air–liquid interface, thereby maintaining alveolar stability. In the absence of surfactant, the work of breathing increases markedly ultimately resulting in respiratory failure secondary to atelectasis, alveolar flooding, and severe hypoxemia. The clinical correlate of surfactant deficiency is the neonatal respiratory distress syndrome (NRDS) in preterm infants, which before the mid-1980s was a devastating and fatal disease. Since the advent of exogenous surfactant replacement therapy, however, newborn mortality from NRDS has decreased approximately 56% from 1987 to 1995.1 The primary surfactant deficiency of NRDS is now a well-characterized condition, and does not appear as complex as the various surfactant changes occurring during acute lung injury (ALI) and/or the acute respiratory distress syndrome (ARDS). Alterations of the endogenous surfactant system in the mature lungs of patients with these disorders are not as well understood, but currently represent an area of intense investigation. This complexity has resulted in inconsistent results of clinical trials evaluating exogenous surfactant administration in this patient population.
This chapter reviews surfactant metabolism and function in the mature lung and its role in maintaining normal lung homeostasis, including its more recently described host defense functions. The metabolism and function of surfactant in the injured lung is also outlined, with particular reference to the effects of mechanical ventilation on the alveolar surfactant system. Subsequently, the status of clinical trials evaluating exogenous surfactant administration in patients with ALI or ARDS is addressed, as are the various factors that may influence a host’s response to this therapy. Future research directions relevant to the understanding of the role of surfactant both in ALI and ARDS and other lung diseases conclude the chapter.
Surfactant Composition and Metabolism
The composition of surfactant is remarkably similar among mammalian species, consisting of approximately 80% phospholipids, 10% surfactant-associated proteins,2–4 and approximately 10% neutral lipids, among which cholesterol is the most abundant (80% to 90% by weight).5,6 The major phospholipid component is phosphatidylcholine, half of which is the disaturated species, dipalmitoylphosphatidylcholine (DPPC).4 This latter molecule is the major surface-active component responsible for lowering surface tension at the air–liquid interface, and is an essential component of all exogenous surfactant preparations currently available for clinical use. Other lipids include phosphatidylglycerol and a few minor lipid species, which are thought to be important in the generation and maintenance of the surface film. The surfactant-associated proteins have been designated as SP-A, SP-B, SP-C, and SP-D.7,8 SP-B and SP-C are small, hydrophobic proteins closely associated with the phospholipids, where they play a major role in generating and maintaining the surface tension-reducing surface film.9–11 SP-B is an 18-kDa dimer, whereas SP-C is a 4-kDa monomer, the latter being the more hydrophobic of these two proteins.12,13 The most clinically effective exogenous surfactant preparations currently in use contain at least one of these natural hydrophobic proteins, or similar types of synthetic and/or recombinant molecules. SP-A is an octodecamer made up of six trimers in a “bouquet” arrangement.14,15 Under reducing conditions, it is a 28-kDa monomer with a 35-kDa glycosylated form. SP-D is a large, multimeric cruciform structure, which is 42 kDa under reducing conditions. Both proteins are very hydrophilic and belong to the collectin family of proteins. They are not components of any of the available natural exogenous preparations consequent to their removal with purification processes, and are not yet available as synthetic or recombinant molecules. Recent evidence suggests that they play a more important role in host defense than in biophysical functions, so there has been renewed interest in developing exogenous preparations containing some form of these proteins.14–17
Surfactant is synthesized within alveolar type II cells (Fig. 60-1).18 Initial assembly occurs in the endoplasmic reticulum with intracellular transport via the Golgi apparatus. Surfactant is stored within lamellar bodies of the type II cell and is secreted into the airspace via exocytosis.19,20 Studies that have investigated the intracellular metabolic pathways of surfactant lipids and proteins have used radiolabeled precursors injected both intravenously and intratracheally. Basically, these studies have shown that although the hydrophobic proteins are assembled and secreted in conjunction with the lipids, SP-A and D are metabolized separately.21 For example, the dominant route for SP-A secretion is by direct, constitutive pathways independent of lamellar body exocytosis, although smaller amounts undergo regulated secretion in association with these organelles.22–24 SP-D is also metabolized independently of surfactant lipids, and, unlike the other surfactant proteins, has been identified in nonpulmonary organs such as the gut.25,26 Within the healthy lung, various pharmacologic agents, such as β-agonists can stimulate surfactant secretion, as can physical stretch of alveolar type II cells.27 This latter phenomenon is particularly relevant to specific aspects of mechanical ventilation because higher tidal volumes increase the stretch of the alveolus, leading to immediate surfactant secretion from type II cells. These effects are relatively short-lived, however, and may be quite different within the injured lung, where the preexisting health of type II cells, before the onset of mechanical ventilation, may be compromised.
Figure 60-1
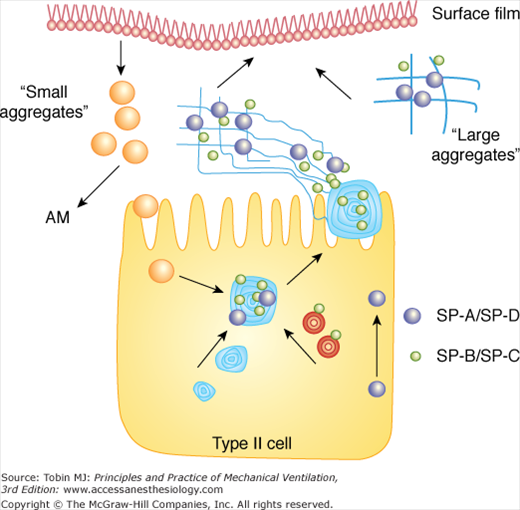
Surfactant metabolism starts with synthesis within the type II cell, secretion into the airspace as lamellar bodies containing both lipid and protein components, and formation of tubular myelin structures. Large aggregates are composed of tubular myelin and freshly secreted lamellar bodies and represent precursors to the surface film. With respiratory motion, small aggregates are formed, which are taken back up into type II cells or cleared via alveolar macrophages (AM).
Recent studies have focused on the extracellular metabolism of pulmonary surfactant, once the material has been secreted from the type II cell into the alveolus (see Fig. 60-1). Within the airspace, mechanical ventilation has a significant and immediate impact on the surfactant system by influencing the metabolism of extracellular surfactant aggregate forms. Because this effect may have important clinical consequences in patients with ALI or ARDS, and is directly affected by different modes of mechanical ventilation, a more detailed discussion of this area is warranted.
Once secreted into the airspace, alveolar surfactant undergoes physical rearrangement from the lamellar structures into tubular myelin, a process involving SP-A, SP-B, DPPC, phosphatidylglycerol, and calcium.28,29 After differential centrifugation of isolated lung lavage, large lipid structures containing SP-A, SP-B, and SP-C, representing the heavier and functionally active forms of alveolar surfactant form a pellet, and are called large aggregates (LA).30,31 They adsorb rapidly to the air–liquid interface and are subsequently converted into smaller, vesicular forms called small aggregates (SA) (see Fig. 60-1). The SA subfractions are poorly functioning, contain little surfactant protein, and are thought to represent surfactant forms that have left the surface film and are subsequently available for reuptake via type II cells for resynthesis into new LA, and/or are cleared entirely from the airspace via catabolism, mainly within alveolar macrophages.32–34
The process of conversion of the functionally active LA forms into inactive SA within the airspace is specifically relevant when discussing the effects of mechanical ventilation on surfactant metabolism. In vitro studies using the surface area cycling technique show that this conversion of LA into SA is mediated by two main factors: a carboxylesterase enzyme called convertase and a phasic change in surface area.31,35–37 The phasic change in surface area factor has been investigated in vivo using different tidal volumes to mechanically ventilate normal rabbits. These studies show that increasing tidal volume, but not positive end-expiratory pressure, results in an increased conversion of LA into SA, and that this conversion occurs relatively soon after the onset of ventilation (Fig. 60-2).38 In normal lungs, these changes do not result in significant alterations in aggregate pool sizes, presumably because of the capabilities of the normal alveolar environment to regulate surfactant metabolism. This does not appear to be the case in the injured lung, however, where alterations in surfactant metabolism occur both intracellularly and extracellularly.39,40 For example, within the lungs of patients with ALI or ARDS, increased aggregate conversion associated with higher tidal volumes is thought to result in increased SA pools, which, together with the associated decrease in functionally active LA pools, may contribute to progressive lung dysfunction. Indeed, these specific changes have been demonstrated in animal models of lung injury and have not only underscored the importance of instituting and maintaining optimal modes of mechanical ventilation within injured lungs, but also in the setting of exogenous surfactant, both during and after administration. This latter situation is discussed in more detail in subsequent sections.
Figure 60-2
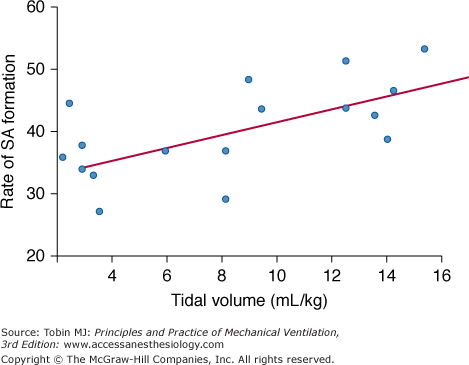
The rate of small aggregate (SA) formation increases in direct proportion to the tidal volumes used to ventilate normal adult rabbits. SA formation was determined by measuring 3H-label recovery in the SA fraction of surfactant 1 hour after injection of a trace dose of 3H-labeled large aggregates into the animals’ lungs. (Reprinted with permission of the American Thoracic Society. Copyright © 2012 American Thoracic Society. From Ito Y, Veldhuizen RA, Yao LJ, et al. Ventilation strategies affect surfactant aggregate conversion in acute lung injury. Am J Respir Crit Care Med. 1997;155(2):493–499. Official Journal of the American Thoracic Society.)
Surfactant Physiology in the Normal Lung
The major role of the surfactant system is related to its biophysical function of lowering surface tension at the air–liquid interface.3,4,41 This is most evident clinically in preterm infants born deficient in surfactant who quickly die of respiratory failure unless supplemented with an exogenous surfactant preparation.42–44 As noted previously, the major lipid component contributing to lowering surface tension is DPPC, although the hydrophobic proteins and some other lipids are also essential.10–13 Full-term babies born deficient in SP-B develop severe lung dysfunction resistant to traditional surfactant therapy and ultimately require lung transplantation for survival.45 Likewise, mice deficient in this protein suffering a similar outcome have also been shown to have abnormal processing of SP-C.46 Mice deficient in SP-C alone, on the other hand, have relatively minor biophysical abnormalities, which predominantly manifest only at low lung volumes, suggesting a potential role for this protein in the surface film stabilization.47
From a physiologic perspective, the initial discovery that surface tension was important for lung stability was made by von Neergaard in 1929, when he observed that it took more pressure to inflate air-filled than saline-filled lungs.48 This was the direct result of the surface tension forces existing at the air–liquid interface. This concept is further advanced by the Laplace law, which states that the pressure gradient across a sphere, which we can roughly extrapolate to an alveolus, is directly related to the surface tension within the sphere divided by the radius of the sphere (ΔP = 2δ/r), where P is pressure, δ is surface tension, and r is radius. From this equation, it is clear that during exhalation, when the alveolar radius decreases, the tendency for this alveolus to collapse increases (i.e., ΔP) unless surface tension decreases as well. Because of its strategic location at the air–liquid interface, and the presence of both hydrophobic and hydrophilic regions, a surfactant film lining the air–liquid interface is able to lower surface tension to near zero levels as the film is compressed at low lung volumes. This decrease in surface tension not only serves to maintain alveolar stability and decrease the work of breathing, but also prevents alveolar flooding because high surface tension tends to draw fluid from the interstitium into the airspace.49 Finally, because surfactant also lines the conducting airways, its biophysical function serves to maintain the patency and stability of small airways and enhance ciliary clearance of particles.50,51 These latter observations suggest that surfactant function may also be relevant in diseases such as asthma, chronic bronchitis, and cystic fibrosis.
As noted previously, the surfactant proteins SP-A and SP-D are members of the collectin family and, secondary to their molecular composition and structure, can bind a variety of bacteria, fungi, allergens, and viruses, thus mediating phagocytosis by immune cells and the killing of pathogens within the lung.14,16,52 In addition, these proteins, as well as some of the hydrophobic components of surfactant, can influence production of nitric oxide, oxygen radicals, and inflammatory mediators from activated cells.53–56 Underscoring the importance of SP-A in host-defense functions are in vivo studies demonstrating that mice deficient in this protein (which are phenotypically normal when not stressed) had increased pulmonary inflammation compared to wild-type animals when bacteria, viruses, or lipopolysaccharide was instilled into their lungs.57,58 These changes were mitigated when exogenous SP-A was administered to these animals. Similar, albeit slightly modified functions of SP-D have been shown both in vitro and in vivo; unlike the normal phenotype of the SP-A null mice, however, the phenotype of unstressed SP-D knockout animals is abnormal with enlarged airspaces and significantly altered surfactant pool sizes.59,60
Although critical for the spreading and stabilization of the surfactant film, SP-B and SP-C also seem to contribute to the host-defense properties of surfactant. Mice deficient in SP-C show greater susceptibility to respiratory syncytial virus infection61 and have increased pulmonary inflammation, decreased macrophage phagocytic activity, and decreased survival compared to wild-type animals following intratracheal instillation of bacteria.62 The role of SP-B in host defense appears to be related to the aminoterminal propeptide of the protein, which improves phagocytosis and has antimicrobial activity at acidic pH.63
The host-defense functions of the various surfactant lipids have also been evaluated. It has been shown that these phospholipids downregulate inflammation and nitric oxide production, presumably because of their “coating” effects on particles and cells.17,64–66 Additionally, surfactant lipids, and DPPC in particular, could have a direct effect in down-modulating the inflammatory response of immune cells, through the regulation of specific molecular pathways responsible for reactive oxygen species production.67 Given the emerging role of the various surfactant components in host defense, future studies involving exogenous surfactant administration may focus more on inflammatory outcomes in addition to biophysical and physiological benefits.
Surfactant Physiology in the Injured Lung
The definitions of ALI and ARDS are rather simplistic and are based on physiologic criteria more than specific etiologies. For example, any acute inflammatory insult that ultimately affects the lungs causing decreased lung compliance, hypoxemia, and bilateral infiltrates on chest radiography (not of cardiac origin) essentially fulfills the diagnostic criteria of ALI and ARDS.68,69 The pathophysiologic changes that occur during this process, including the surfactant alterations reported in numerous studies, are complex and reflect the lack of effective treatments for this disorder.
The first postmortem descriptions of the lungs of patients dying from ARDS suggested that surfactant dysfunction may have played a role in their demise.70 Subsequently, many studies have reported consistent changes in the surfactant system in lavage samples obtained from these patients, including decreased phosphatidylcholine and DPPC levels, decreased phosphatidylglycerol levels, decreased SP-A, SP-B, SP-C, and SP-D levels, and a decrease in the functionally active LA forms relative to SA.71–73 Interestingly, in these severely ill patients, serum levels of SP-A and SP-D were shown to be elevated, likely consequent to the associated marked increase in pulmonary permeability in this setting.74,75 The biophysical consequence of these collective changes in the endogenous surfactant system is an inability to adequately reduce surface tension, resulting in decreased lung compliance, increased permeability with edema formation, and hypoxemia. The next section addresses the mechanisms responsible for the surfactant changes observed in these patients. Only with a better understanding of these mechanisms will optimal treatment strategies be developed that are aimed at restoring normal surface tension forces within injured lungs, or perhaps, more importantly, preventing the development of these changes.
Unlike NRDS, in which the preterm animal represents a reasonable correlate of the clinical condition of NRDS and is thus suitable for reliable mechanistic and therapeutic studies, no one animal model adequately reflects the complexity of patients with ALI or ARDS. Proposed mechanisms in the latter setting are, therefore, derived from many different animal models, a factor that needs to be considered when extrapolating results of such studies to the clinical situation.
The observed changes in the phospholipid composition of endogenous surfactant within the injured lung represent a relatively late finding in models of ALI when severe lung dysfunction is present. These changes are likely related to abnormal synthetic and/or secretory pathways within the type II cell, as well as alterations in the degradation process of some lipids within the airspace via increased phospholipase activity (Fig. 60-3).76–79 A similar mechanism is likely responsible for the decreased surfactant protein levels observed in these lungs, although with the permeability abnormalities demonstrated in these patients, there may also be transfer of SP-A, SP-B, and SP-D across the alveolar–capillary barrier into the serum.74,75 Interestingly, clinical studies show that serum levels of SP-A and SP-B are inversely related to oxygenation values in patients with ARDS, and serum SP-D levels positively correlate with the survival of these patients.74,75 In addition to these quantitative changes in surfactant protein levels, there may also be modifications to their composition in the form of nitration and/or oxidation, which would also compromise their function but may not impact measurable levels.80–82
Figure 60-3
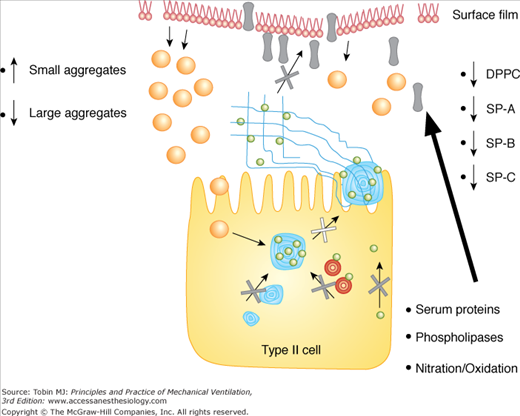
Mechanisms leading to surfactant alterations in the injured lung include abnormal type II cell metabolism, increased phospholipase activity, nitration and oxidation of surfactant, increased conversion of large aggregates into small aggregates, and inhibition of surfactant by serum proteins leaking into the airspace. See text for details.
The mechanisms responsible for the changes observed in surfactant aggregate forms were alluded to earlier. Briefly, decreased LA pools may be caused by decreased type II cell synthesis and/or secretion in the severely injured lung, but an increased conversion of LA into SA forms likely occurs at earlier stages of the disease, particularly in patients undergoing mechanical ventilation.37–40 This was recently investigated in animal studies in which spontaneously breathing septic rats with relatively mild lung dysfunction had unchanged LA pools, whereas SA pools were significantly decreased compared to sham animals.83 These changes were opposite to those documented in severely injured lungs; it was hypothesized that they may have represented the host’s compensatory response aimed at maintaining endogenous surfactant in LA forms. For example, by utilizing smaller tidal volumes (with higher respiratory rates), these animals would decrease LA conversion secondary to the smaller changes in alveolar surface area, thereby attempting to maintain lung function. Indeed, very soon after the onset of mechanical ventilation of these animals, which involved using higher tidal volumes than those associated with spontaneous breathing, conversion of LA into SA increased dramatically and lung function deteriorated rapidly.84 Of clinical relevance, however, was the ability to mitigate both the aggregate conversion and progressive lung dysfunction observed by using lower tidal volumes from the onset of ventilation. In fact, high-frequency oscillation (HFO), a strategy involving extremely small tidal volumes with high respiratory rates, proved to be superior to all other “lung-protective” modes of ventilation with respect to aggregate conversion, lung function, and inflammatory changes.85 Interestingly, these laboratory results are consistent with recent clinical trials evaluating different modes of mechanical ventilation in patients with ALI. One large trial showed that lower tidal volumes resulted in superior outcomes compared to higher tidal volumes in patients with ALI or ARDS.86 Results of phase III clinical trials evaluating the outcome of patients specifically ventilated with HFO are pending.
Recent studies suggest another potential mechanism for surfactant dysfunction within the injured lung. Alterations in the cholesterol-to-phospholipids ratio of surfactant alter surfactant function. As noted earlier, cholesterol is the major neutral lipid in surfactant and when present in physiologic amounts, it enhances adsorption at the air–liquid interface. Studies focused on the specific role of neutral lipids and cholesterol in lung injury are scarce. In vitro studies that have altered the cholesterol-to-phospholipid ratio show that supraphysiologic levels of cholesterol affect surfactant film stability leading to the inhibition of its function.87 This cholesterol-mediated dysfunction appears to be relevant in vivo as well, because surfactant isolated from animals exposed to high tidal-volume ventilation showed abnormal biophysical activity, which was subsequently restored upon cholesterol removal.88
Interestingly, these experimental results are consistent with clinical observations. Markart et al showed that patients with ARDS had increased levels of the neutral lipid component in isolated LA forms, and impaired biophysical activity of their endogenous surfactant.89 The source of the increased cholesterol in the surfactant of injured lungs is currently unknown. Altered synthesis by alveolar epithelial type II cells and/or leakage from the circulation into the alveolus are potential mechanisms, although this has not been formally evaluated. Further studies are necessary to address these issues, as well as to understand the biophysical mechanisms leading to cholesterol-mediated inactivation of surfactant.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


