CHAPTER 25 Sudden Cardiac Death
Definition
Sudden cardiac death (SCD) is defined as a natural and unexpected death as a result of cardiac causes that occur within 1 hour of the onset of new symptoms.1 In some medical circles, the term “sudden cardiac arrest” (SCA) is preferred because the expression “death” conveys finality of the event, which is not necessarily the outcome in all cases. In this communication, both terms will be used as appropriate in the text.
Epidemiology
Cardiovascular disease-related deaths are still the most common cause of mortality in the United States. The two modes of cardiovascular death (i.e., sudden and nonsudden) are equally common. SCD claims 325,000 lives each year in United States,2 thus the incidence is 0.1% to 0.2% annually.3 It is usually ascribed to arrhythmic causes and it is the case the vast majority of the time when the initiation of the episode is electrocardiographically documented. However, the victims are seldom under medical observation, thus the exact mechanism leading to cardiovascular collapse is difficult to establish, and the cause is labeled on the basis of presentation and the earliest onsite ECG recordings. Monitored victims and rescue squads encountering individuals with SCA show the following arrhythmias at the time of arrival: ventricular fibrillation (VF) in 80% of the victims, ventricular tachycardia (VT) in 10% who have the best outcome, and bradycardias in 10%.4,5 The rhythm disturbance first documented by emergency rescue teams is dependent upon the time elapsed after the cardiovascular collapse. VF is seen early after collapse and progresses to asystole as time passes (Fig. 25-1).
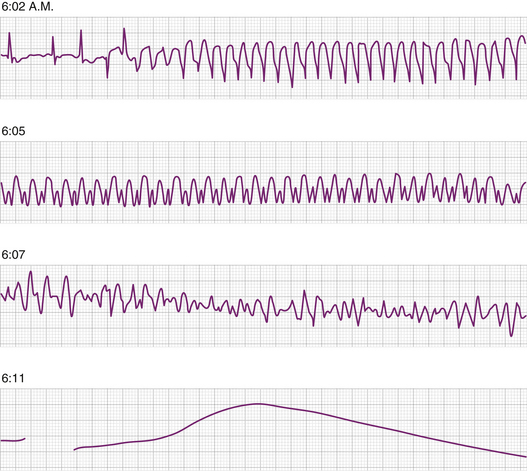
For the most part, patients with bradycardia have little or no response to pacing, suggesting hemodynamic collapse as the underlying problem and bradycardia as the concomitant rhythm (electrical-mechanical dissociation). Occasionally, torsades de pointes may be precipitated by severe bradycardia and cause SCA. The universal futility of cardiac pacing in this setting to improve survival in this population, attests to the possibility that overall, one fourth of SCA victims may have nonarrhythmic etiology. Conduction system abnormalities have been labeled as the cause of SCD in younger population victims without demonstrable heart disease in autopsy studies.6
Pathophysiology
Pathologic Substrates
Over the years, numerous studies have revealed that an overwhelming majority of SCA cases (75% to 80%) are due to acute, chronic, or acute on chronic atherosclerotic CAD.1 The second most frequent category of disorders is cardiomyopathies, which include both hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM). These pathologic substrates are:
Functional Modulators
Clinical Substrate
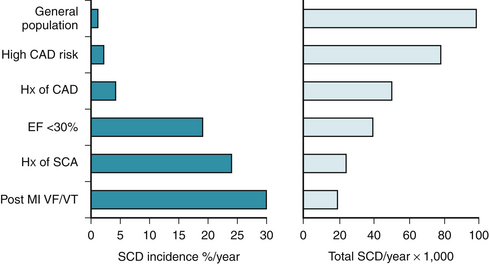
To predict SCA, it is important to recognize the conditions leading to abrupt cessation of cardiac output. Figure 25-2 shows data derived from various studies22–26 demonstrating the predominant substrates of SCD. The relative risk for SCD is dependent upon the underlying substrate and is graphically demonstrated for various populations in Figure 25-3.
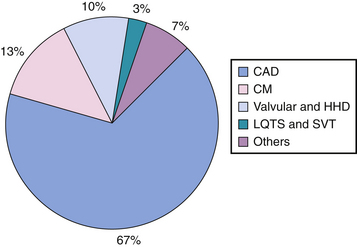
Figure 25-2 Prevalence of underlying heart disease in adult patients who have experienced sudden cardiac death, based on data derived from several studies,22–26 shows coronary artery disease, cardiomyopathies, valvular and hypertensive heart disease, and electrical disturbances as the predominant substrates. CAD, coronary artery disease; CM, cardiomyopathy; HHD, hypertensive heart disease; LQTS, long QT syndrome; SVT, supraventricular tachycardia.
(From Deshpande S, Vora A, Axtell K, Akhtar M: Sudden cardiac death. In Brown DL [ed]: Cardiac Intensive Care. Philadelphia, Saunders, 1998, pp 391-404.)
Coronary Artery Disease (CAD)
CAD is the most common cause of SCA, where it is the responsible substrate in 65% to 90%27–33 of cases. SCA related to coronary events is more common in younger victims, more common in men than in women, and more common in African Americans than in whites. Approximately 20% of SCA victims show evidence of new ST elevation myocardial infarction at the time of cardiac arrest, but 40% to 75% have evidence of healed MI. Fifty percent of post–MI deaths occur in the first 6 months.9 Most of the risk factors for CAD are predictors for SCA (e.g., left bundle branch block on ECG, hyperlipidemia, hypertension, cigarette smoking, obesity, diabetes, and lifestyle).3
Nonatherosclerotic CAD
Hypertrophic Cardiomyopathy (HCM)
Despite a relatively low incidence of HCM in the general population, it maintains a high profile risk because 50% to 90% of deaths in patients with HCM are sudden, with an annual mortality rate of 2% to 4%.45 Unlike most other heart diseases, the risk of SCA in HCM declines with age.46 Patients with this disease characteristically have genetic heterogeneity in the amount of hypertrophy in different regions in the left ventricle. It results from multiple diseased genes, encoding sarcomeric proteins. Microscopically, gross disorganization of muscle bundles and myofibrillar architecture, altered gap junctions, increased basal membrane thickness, and connective tissue accumulation are noted.47 These patients, therefore, manifest both electrical instability and consequences of myocardial hypertrophy with altered hemodynamics.
The mortality risk, based on the degree of outflow tract obstruction, has not yet been clearly stratified and, therefore, although hemodynamic considerations may play an important role in SCA, no predictive variables have been identified.47 Furthermore, the combination of nonsustained ventricular tachycardia (NSVT) and inducible VT at electrophysiologic testing does confer a higher risk, and, conversely, the absence of NSVT and lack of inducibility indicates a lower risk.
The mechanism for SCA in patients with HCM is most often ventricular arrhythmias.48 However, left ventricular outflow obstruction, sinus node dysfunction, atrioventricular (AV) conduction diseases, and supraventricular arrhythmias are alternative mechanisms.49 The genesis of these arrhythmias is found in a complex interplay of electrophysiologic and hemodynamic abnormalities, primarily from electrophysiologic derangement of the hypertrophied muscle. Fairly often, SCA is the first manifestation of heart disease in these individuals, and thus is implicated as the substrate explaining the occurrence of these arrhythmias in children or young athletes.50 Young age, strong family history, worsening obstructing symptoms,51 prior episode of SCA, failure to raise blood pressure on exercise, and severe ventricular hypertrophy of more than 3 cm thickness52 are incremental risk factors for SCA.
Ventricular hypertrophy secondary to systemic or pulmonary hypertension or owing to valvular or congenital heart disease is also associated with an increased risk for SCA.11 This risk is proportionate to the level of severity of the hypertrophy.53
Idiopathic Dilated Cardiomyopathy (DCM)
Overall survival following a clinical diagnosis of DCM is estimated to be 70% at 1 year and 50% at 2 years.54 Most of these deaths are sudden in nature, and the incidence ranges in various series from 28% to 72% of total deaths. Syncope is a poor prognostic indicator in patients with DCM and is associated with a 44% incidence of SCD in 4 years.55 Patients with preserved left ventricular function may have a lower short-term mortality from pump failure but are at a greater risk for SCA from arrhythmic events. Ventricular tachyarrhythmias56 are the most common mode of death, but bradyarrhythmias57 occur also, especially in those patients with advanced pump dysfunction. The arrhythmia most commonly implicated in SCA is primary polymorphic VT or VF. Additionally, rapid sustained monomorphic VT, related to bundle branch reentry,58 and monomorphic VT, unrelated to bundle branch re-entry, have been seen. The recognition of this arrhythmia is critical because these patients can be successfully cured by catheter ablation of the right bundle branch.58 Catheter ablation, at the present time, cannot be considered a curative option in SCA survivors with ventricular tachycardia related to CAD. Monomorphic VT, unrelated to bundle branch re-entry, is probably the result of the presence of smaller re-entrant circuits within the myocardium. In most instances, the triggering mechanism for the onset of primary polymorphic VT or VF is unclear. In some patients, triggers such as hypokalemia, use of antiarrhythmic medications, and hypomagnesemia may be more easily identifiable.
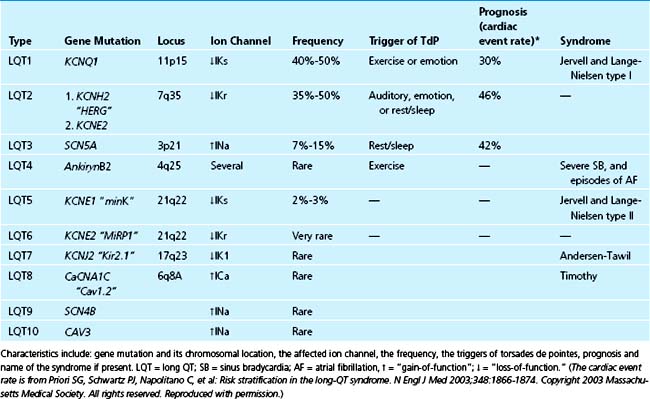
Long QT (LQT)
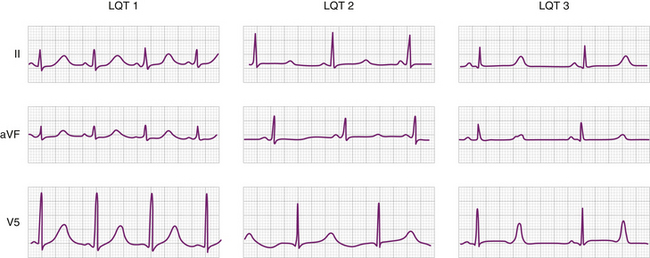
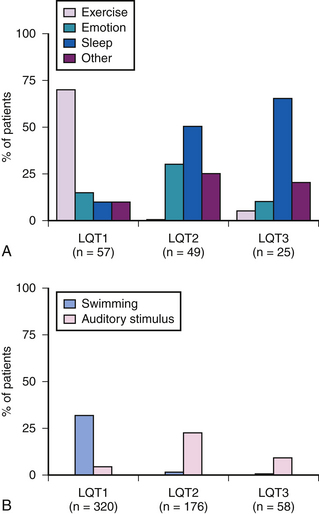
Brugada Syndrome
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
 Hemodynamically Unstable Presentations of Congenital Heart Disease in Adults
Hemodynamically Unstable Presentations of Congenital Heart Disease in Adults
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Antiplatelet Therapy
Antiplatelet Therapy
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


