Chapter 57 Structure, Function, and Development of the Nervous System
Major Cell Types
Glial cells are typically divided into microglia and macroglia. Microglia are phagocytic cells derived from peripheral macrophages, and normally exist in a resting, or quiescent state in the central nervous system (CNS). Microglia are activated by several physiologically relevant factors, including bacterial lipopolysaccharide in the setting of infection and thrombin in the setting of injury.1 Whether microglial activation is neurotoxic or neuroprotective in these settings has yet to be fully characterized. In addition, activated microglia play an important role in the neuropathology of Alzheimer disease, HIV-associated dementia, and prion diseases. Therapeutic strategies targeting microglial activation are just now beginning to emerge in animal disease models2–3 and human clinical trials.4
Intercellular Communication in the Nervous System
Electrical Synapses
At the electrical synapse, cell membranes of the adjoining neurons are tightly bound together into a gap-junction plaque.5 Each plaque contains numerous channels made of connexin proteins. There are 21 known connexin genes in humans. Each channel consists of two hemichannels, with one on each cell membrane. Two hemichannels join together to form a functional gap junction between two neurons, allowing intercellular diffusion of ions and small molecules such as glucose, cyclic AMP, and ATP. Gap junctions thus allow neurons to share information about their metabolic and excitable states, providing a mechanism for large-scale regulation of energy demands and neuronal network dynamics. Additionally, gap junction channels close in response to lowered intracellular pH or elevated Ca2+ levels; since both events occur in damaged cells, paired hemichannels at the gap junction may function to isolate healthy neurons from those damaged during ischemia or trauma. Recent evidence suggests that unpaired hemichannels outside of the gap junction plaques may also contribute to ischemic neuronal cell death.6
Glia, like neurons, are also connected by gap junctions. For example, brain astrocytes form an interconnected cellular network, which allows long-distance propagation of calcium signals across many cells. Additionally, layers of myelin generated by oligodendrocytes in the CNS and by Schwann cells in the PNS are linked by gap junctions. Myelin gap junctions provide structural stability to the myelin sheath and allow for rapid diffusion of nutrients and other substances across the sheath towards the underlying axon. In humans, mutations in gap junction protein connexin 32 result in X-linked Charcot-Marie-Tooth disease, a demyelinating neuropathy.7
Chemical Synapses
Neuromuscular Junction
The NMJ consists of three distinct anatomic components: the presynaptic nerve terminal, the synaptic cleft containing the basement membrane, and the postsynaptic muscle fiber. The presynaptic nerve terminal originates from the myelinated axon of a motoneuron in the spinal cord. Lower motoneurons in the ventral gray matter of the spinal cord send their myelinated axons through the ventral root towards peripheral muscle targets. As the axon approaches the muscle fibers, it loses its myelin sheath and branches into a fine network of terminals, each approximately 2 μm in diameter. Each branch has several swellings along its course, termed presynaptic boutons, where the nerve makes synaptic contact with the muscle fiber. Presynaptic boutons are covered by terminal Schwann cells, which provide growth factors, recycle neurotransmitters, and may participate in recovery after nerve and muscle injury. Presynaptic boutons are positioned over specialized regions of the muscle cell membrane called the end-plate regions. Furthermore, underlying each bouton is a specialized invagination of the cell membrane that contains a very high concentration of nicotinic acetylcholine (ACh) receptors and voltage-gated Na+ channels. The presynaptic bouton and the end-plate are separated by a 100 nm-wide synaptic cleft containing the basement membrane and the extracellular matrix. The basement membrane anchors a number of proteins, including acetylcholinesterase, the enzyme responsible for rapid hydrolysis of ACh in the synaptic cleft.
A detailed understanding of diseases that affect synaptic transmission at the NMJ, as well as familiarity with clinical pharmacology as it applies to the NMJ, are essential in critical care. Specific diseases affecting the NMJ include toxin-mediated botulism and autoimmune disorders such as myasthenia gravis, Lambert-Eaton syndrome, and neuromyotonia. These are discussed in detail in Chapter 64 (see also Tseng-Ong and Mitchell8 and Lang and Vincent9). Among the pharmacotherapies targeted at the NMJ are some of the most commonly used drugs in the pediatric ICU: neuromuscular blockers (NMB). NMB agents are reviewed in Chapter 122. Furthermore, a number of toxins either decrease (anticholinergic agents) or increase (acetylcholinestrase inhibitors) the amount of ACh available at cholinergic synapses, resulting in corresponding toxidromes (see Chapter 106).
Neurotransmitter Systems
Several substances are employed for communicating information chemically between neurons in the nervous system or between a neuron and a muscle at the neuromuscular junction. These substances are called neurotransmitters, and generally fall into three categories: amines, amino acids, and peptides (Table 57-1). Each neurotransmitter requires its own synthetic machinery and exerts specific actions on the postsynaptic target. Furthermore, neurons tend to be characterized anatomically, immunohistochemically, and functionally by the main neurotransmitter they use, allowing insight into the role of each neurotransmitter system in CNS function.
Table 57–1 Neurotransmitter Classes
| Amines | Amino Acids | Peptides |
|---|---|---|
| Acetylcholine | γ-Aminobutyric acid | Substance P |
| Dopamine | Glutamate | Vasoactive intestinal peptide |
| Norepinephrine | Glycine | Dynorphin |
| Epinephrine | Enkephalins | |
| Serotonin | Neuropeptide Y | |
| Histamine | Cholecystokinin |
Neurotransmitters operative exclusively in the central nervous system are in normal font, those used both in the central and peripheral nervous systems are in italics, and those used exclusively in the peripheral nervous system are in bold.
Neurotransmitters
Acetylcholine
Acetycholine is an amine that functions as a neurotransmitter at the NMJ as well as in the CNS. At the NMJ, its actions are quick and precise, whereas in the CNS it functions as a slow, more global modulator of synaptic activity. Cholinergic CNS neurons include all the motoneurons in the spinal cord, as well as a number of cell nuclei in the brainstem reticular formation and the basal forebrain. Cholinergic neurons synthesize ACh using choline acetyltransferase (ChAT), which transfers an acetyl group from acetyl coenzyme A (CoA) to choline (Figure 57-1, A). Choline concentration in the extracellular fluid is the rate-limiting step in the reaction. Cholinergic neurons also synthesize acetylcholinesterase (AChE), the enzyme that breaks down ACh. AChE is released with ACh into the synaptic cleft, where it rapidly hydrolyzes ACh into acetic acid and choline.
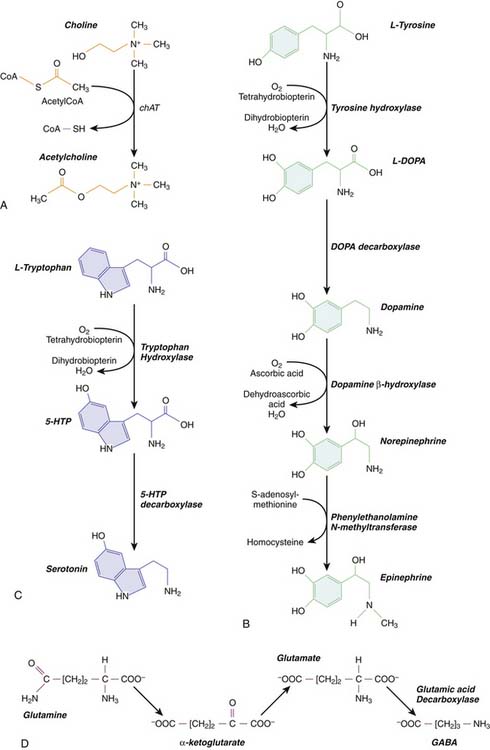
Low concentrations of choline in the CNS have been associated with neurologic impairment during fetal10 and postnatal life.11 Thus, choline supplementation represents an attractive therapeutic strategy in neurologic disorders characterized by decreased CNS choline. Interestingly, patients receiving long-term parenteral nutrition (TPN) occasionally develop choline deficiency, which has been associated with TPN-related liver failure12 and possibly, cognitive dysfunction. Hence, choline supplementation in TPN-dependent patients may ameliorate some neurologic deficits.13
Catecholamines
Catecholamine neurotransmitters include dopamine, norepinephrine and epinephrine. For all three, the initial starting point in biochemical synthesis is the amino acid tyrosine (Figure 57-1, B). Tyrosine is converted into an intermediate compound, called dopa, by tyrosine hydroxylase (TH). In dopaminergic neurons, dopa is converted into dopamine by dopa decarboxylase. Noradrenergic neurons, which utilize norepinephrine as a neurotransmitter, further convert dopamine into norepinephrine using dopamine β-hydroxylase. Finally, norepinephrine is converted into epinephrine by an enzyme phentolamine N-methyltransferase, which is found only in adrenergic neurons. Thus all neurons that synthesize catecholamines contain TH and dopa decarboxylase, but only noradrenergic and adrenergic neurons contain the synthetic enzymes required to produce norepinephrine and epinephrine, respectively.
Serotonin
Serotonin is an amine neurotransmitter synthesized from the amino acid tryptophan in a two-step process (Figure 57-1, C). First, tryptophan is hydroxylated by tryptophan hydroxylase to form 5-hydroxytryptophan (5-HTP). 5-HTP is then decarboxylated by 5-HTP decarboxylase to form serotonin, also known as 5-hydroxytryptamine (5-HT). After 5-HT is released at the synapse, it is cleared by a specific serotonin reuptake transporter. Serotonergic neurons are located in the rostral and caudal raphe nuclei in the brainstem. Rostral raphe neurons innervate the cerebral cortex, including the limbic system, where serotonin levels help regulate mood and attention. Caudal raphe neurons project to the brainstem and the spinal cord, where they are involved in regulation of general arousal and pain perception, respectively. Importantly, dysfunction of the serotonergic pathways originating in the raphe nuclei has been linked with sudden infant death syndrome (SIDS).14 Additionally, serotonin levels play a key role in depression, giving rise to an entire class of drugs in clinical use called selective serotonin reuptake inhibitors (SSRIs). SSRI abuse or overdose is rare, but may result in patients presenting with the potentially life-threatening “serotonin syndrome,” characterized by hypertension, tachycardia, mental status changes, myoclonus, and severe hyperthermia. The latter may lead to shock, rhabdomyolysis, renal failure, and death. The serotonin syndrome is particularly likely to occur when SSRIs and MAO inhibitors, inadvertently or intentionally, are taken together. Treatment includes serotonin antagonists, blood pressure control with either adrenergic antagonists or agonists as clinically indicated, and temperature control with benzodiazepines and neuromuscular blockade.
Amino Acids
Neurotransmitters derived from common amino acids include glutamate, gamma-aminobutyric acid (GABA), and glycine. These are among the most widely distributed neurotransmitters in the CNS. Glutamate and glycine exist as amino acids in all cells, where they are utilized as protein building blocks. Glutamatergic and glycinergic neurons have the additional capacity to package glutamate and glycine, respectively, into synaptic vesicles and release them at the synapse. GABA must be synthesized from glutamate via an additional reaction catalyzed by an enzyme glutamic acid decarboxylase (GAD) (Figure 57-1, D). Only GABA-ergic neurons contain GAD.
Glutamate is generally an excitatory neurotransmitter, whereas GABA and glycine are inhibitory. Excitatory glutamatergic neurons exert their influence both locally and over long distance, depending on the shape of their axons. Inhibitory neurons, on the other hand, tend to exert local inhibitory control over neuronal circuitry either in the brain (GABA) or in the spinal cord (glycine). A major exception are cerebellar Purkinje cells, which are GABA-ergic but project over long distances to the brainstem, thalamus, and cerebral cortex (see below).
Adenosine, Peptides, and Nitric Oxide
Finally, several gaseous molecules function as neurotransmitters. These include nitric oxide (NO) and carbon monoxide (CO), and possibly hydrogen sulfide. NO, the most thoroughly studied of the gaseous neurotransmitters, is produced by brainstem neurons in the nucleus tractus solitarius, where it interacts with α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)-type and N-methyl-D-aspartate (NMDA)-type glutamate receptors and regulates cardiovascular function.15 Hydrogen sulfide is produced from the amino acid cysteine, and may influence cellular redox state and glutamatergic transmission.16 Intriguingly, hydrogen sulfide induces a suspended animation-like state in animals.17 and may be protective after resuscitation from cardiac arrest.18
Neurotransmitter Receptors
Nicotinic Acetylcholine Receptors
Nicotinic ACh receptors are ligand-gated ion channels, related structurally and functionally to GABAA channels, and a subset of serotonin receptors. Five transmembrane subunits comprise the nAChR and form a central pore that allows ionic currents to pass. There are two subunit classes, α and β, with multiple members in each class. nAChR is generally a heteromer, but homomer channels have been described. Each nAChR binds two acetycholine molecules, with affinity for ACh and for nicotine dependent on subunit composition. The receptor exists in three distinct states: closed, open, and desensitized. In the closed position, no ionic current passes through the central core. When an agonist, such as ACh or nicotine, binds the nAChR, the receptor opens, becoming permeable to monovalent and divalent cations. After a short period of time, the receptor spontaneously closes. Upon continued exposure to an agonist, however, the nAChR assumes a permanently closed, or desensitized, conformation.19 As discussed above, nAChRs mediate neuromuscular coupling at the NMJ. In addition, nAChRs are widely distributed in the CNS, where they perform a variety of functions depending on subunit composition and location.
In the CNS, unlike at the NMJ, nAChRs are permeable to both Na+ and Ca2+. In neurons, however, nAChR-evoked Ca2+ current exceeds the Na+ current twofold to tenfold. The greater Ca2+ permeability indicates that nAChRs mostly modulate synaptic transmission and release of other neurotransmitters. Indeed, although direct nAChR-dependent responses have been observed in the hippocampus and the developing visual cortex, the overwhelming evidence points to nicotinic receptors playing the role of modulator in the CNS. Their wide distribution in the CNS with locations presynaptically, postsynaptically, and extrasynaptically further supports that role. Activation of presynaptic nAChRs enhances release of ACh, dopamine, glutamate, and GABA. Coupling of enhanced glutamate release with nAChR-dependent increase in intracellular Ca2+ suggests that nAChRs participate in synaptic plasticity during learning. Postsynaptic and extrasynaptic nAChRs regulate excitability and signal propagation in neuronal circuits. In the hippocampus, for example, nAChR activation leads to increased release of GABA from inhibitory interneurons, which decreases the excitability of hippocampal pyramidal neurons. Nicotinic receptors also interact with the dopaminergic system in regulating neuronal circuitry in the basal ganglia and the limbic system. Thus, nicotinic receptors have been implicated not only in learning and memory, but also in regulation of addiction and reward. Furthermore, loss of cholinergic neurons represents one of the distinguishing neuropathologic features of Alzheimer disease, and cholinesterase inhibitors are widely used to improve cognition and memory in Alzheimer patients.19 In pediatrics, a mutation in nAChR is responsible for a specific clinical epilepsy phenotype, called autosomal dominant nocturnal frontal lobe epilepsy.20 Seizure onset usually occurs around 12 years of age in otherwise healthy children. Seizures originate in the frontal lobe, and occur predominantly during non-REM sleep.21 The mutant nAChR is more sensitive to ACh than the wild-type receptor, suggesting that cholinergic medications should be avoided in these patients.
Muscarinic Acetylcholine Receptors
Muscarinic ACh receptors (mAChRs) comprise a group of metabotropic receptors that link ACh exposure at the surface with G protein activation inside the cell. There are five distinct subtypes of mAChRs, designated M1 through M5, and these subtypes are divided into two broad classes based on the identity of the G protein with which they interact. M2 and M4 mAChRs couple with Gi proteins, and inhibit adenylyl cyclase activity and reduce intracellular cAMP levels. M1, M3, and M5 receptor subtypes couple with Gq proteins, and increase intracellular Ca2+ levels via activation of phospholipase C.22 In the CNS, the M1 mAChR is the most abundant subtype, located on neurons in the cortex, thalamus and striatum.23 mAChRs are also present in the PNS in the sweat glands and organs of lacrimation and salivation, as well as in the heart, where they mediate the parasympathetic control of heart rate and contractility. Muscarinic AChRs thus mediate many of the systemic effects of organophosphate exposure and nerve gas poisoning.24
Glutamate Receptors
Glutamate is the major excitatory neurotransmitter in the central nervous system. In mammals, it depolarizes postsynaptic neurons by binding to three types of ionotropic glutamate receptors (iGluR), each of which is characterized by different affinities for synthetic analogues, by different selectivity to ions, and by different time-course of the current that is permitted to pass through the cell membrane. The three types of iGluR are the AMPA receptor, the NMDA receptor, and the kainate receptor. All three channels types are widely present throughout the CNS, with AMPA and NMDA channels mediating the bulk of the excitatory transmission. At present, the function of the kainate channel remains to be clearly defined.
AMPA and NMDA receptors differ from each other with respect to ion permeability and time-course of ion flow through the channel. Upon binding of glutamate, AMPA receptors open their pores, which are permeable to Na+ and K+ ions. At the negative resting potential of the neuronal cell membrane, Na+, driven by the electrochemical gradient, flows into the cell and causes a large, fast depolarization. AMPA receptors are generally impermeable to Ca2+, although more recent findings indicate that Ca2+-permeable AMPA receptors do exist and may significantly contribute to pathology observed in ALS and stroke.25 In contrast, NMDA receptors are universally permeable to Ca2+, as well as to Na+ and K+, generating a slow inward depolarizing current. NMDA receptors possess a unique property, however, that allows them to pass current only when the neuronal membrane is already depolarized. This property, termed voltage-dependence, stems from Mg2+ ions blocking the entry pore of the NMDA channel at negative membrane potentials even in the presence of glutamate. As the neuron depolarizes further, mostly due to current flow via the AMPA receptor, the Mg2+ block is relieved, and Ca2+, as well as Na+, flow into the cell. Once inside the cell, Ca2+ ions mediate a multitude of effects, from modifying protein phosphorylation and gene expression to overt excitotoxicity and cell death. NMDA receptors are thus thought to function as coincidence detectors, linking events at the cell membrane, e.g., AMPA receptor-mediated depolarization, with long-term changes in synaptic strength and gene expression in the neuron.
In addition to directly modulating current flow via the ionotropic channels, glutamate modulates effector molecules within the neuron by binding to a diversity of G protein-linked metabotropic glutamate receptors (mGluRs). Eight mammalian subtypes of mGluR are divided into three categories, based on sequence homology and coupling to secondary effector systems.26 Group I mGluRs (mGlurR1 and mGluR5) are localized at the edge of the postsynaptic neuronal membrane and are positively coupled to phospholipase C (PLC) via the Gq protein. Group II and III mGluRs are located on the edge of the presynaptic neuronal membrane and are negatively coupled to adenylyl cyclase (AC) via the Gi protein. All three groups are activated only when excess glutamate spills out of the synaptic cleft and diffuses towards mGluRs located at the periphery of the synaptic membrane.
In contrast, group III, and probably group II, mGluRs on presynaptic neurons provide a negative feedback loop by inhibiting glutamate release. When glutamate binds to group III mGluRs, an inhibitory G protein (Gi) is activated, that then functions to decrease AC-mediated production of cAMP. A decrease in cAMP leads to lower Ca2+ concentrations at the presynaptic neuronal membrane and decreased synaptic vesicle fusion. The net effect is a decrease in the amount of glutamate released at the synapse and a reduction in synaptic transmission. Recently, mGluRs have emerged as a major therapeutic target due to the multitude of effects they exert on synaptic transmission. Their extrasynaptic location presumably will allow newly developed pharmaceutical agents to maximize therapeutic value and minimize unwanted side effects.27
GABAA and GABAB Receptors
GABAA receptors are heteropentamers, similar in structure to the nAChRs. At least eight subunit classes exist in mammals, including humans, and each class consists of several members, allowing for a staggering 150,000 possible subunit combinations to create one functional GABAA receptor. Only 500 combinations are known to exist, and most receptors contain a various complement of the α, β, and γ subunits.28 Most GABAA receptors cluster at postsynaptic densities, and such clustering appears to depend on presence of the γ subunit. However, a subset of the GABAA receptors, in particular those containing the δ subunit, localize to extrasynaptic sites, mediate tonic levels of inhibition in the brain, and may underlie the pathophysiology of absence seizures.29
The pharmacology of the GABAA receptor is of particular relevance in critical care, since the two classes of first-line anticonvulsants and anxiolytics in clinical practice—the benzodiazepines and the barbiturates—allosterically modulate the GABAA receptor. GABA itself binds the receptor at the junction of the α and β subunits. Benzodiazepines bind the GABAA receptor at a different site, classically between the α and γ2 subunits, and, in the presence of GABA, increase the frequency with which the chloride channel opens. Barbiturates, in contrast, bind at yet a different site and increase the duration of the open state in the presence of GABA. Thus both benzodiazepines and barbiturates increase the efficacy of endogenous GABA in hyperpolarizing the cell membrane. A major difference between the two drug classes is that at increasing concentrations, barbiturates, but not benzodiazepines, become direct GABA agonists and can open GABAA channels independent of endogenous GABA release. Hence, barbiturates have a significantly narrower safety window compared to benzodiazepines.
GABAA receptors mutations contribute to several known disease states in humans. Two different point mutations on chromosome 5, both affecting the γ2 subunit, are associated with development of febrile seizures and generalized epilepsy as well as with a link between the two conditions.30–31 In patients with temporal lobe epilepsy, GABAA receptor expression is altered in hippocampal neurons,32 and GABA-evoked responses actually depolarize, rather than hyperpolarize, neurons in excised tissue (see also Developmental Processes section later in the chapter).33 GABAA receptor dysfunction is also thought to contribute to anxiety, panic disorder, schizophrenia, and sleep disturbances.34 Thus, pharmacologic modulation of GABAA receptor function represents an active area of research and novel drug development.
GABAB receptors are heterodimeric proteins that activate G protein-coupled second messenger systems upon interaction with GABA at the membrane.35 The GABAB1 subunit binds GABA at the cell membrane, while the GABAB2 subunit interacts with the G protein on the cytosolic surface. GABAB receptors are widely distributed throughout the CNS but are particularly abundant in the thalamus, cerebellum, and hippocampus. They can be located both presynaptically and postsynaptically. Presynaptic GABAB receptors allow G protein–coupled Ca2+ influx into the cell, and leads to feedback inhibition of transmitter release. Postsynaptic GABAB receptors function as K+ channels by allowing a slow, G protein–dependent K+ current to leak out of the cell, and leads to cell hyperpolarization (since positive ions have left the inside of the cell membrane).36
Baclofen is the only pharmacologic GABAB receptor ligand in current clinical use. It directly activates the GABAB receptor and, through its action in the spinal cord, leads to reduction in muscle tone. Thus it is used primarily to relieve spasticity after CNS injury. When administered systemically, it has a significant side-effect profile, including hypotension and bradycardia.35 Systemic side effects have been minimized with intrathecal administration via an indwelling catheter and pump.37,38 Indeed, intrathecal baclofen infusion has emerged as an alternative to the more invasive dorsal rhizotomy in the treatment of spasticity refractory to medical therapy in pediatric patients.39 Although intrathecal baclofen delivery systems are effective, they have a 10% to 20% failure rate over time. When an intrathecal baclofen pump fails, patients can develop baclofen withdrawal symptoms characterized by agitation, increasing spasticity and dystonia, hypertension, tachycardia, hyperthermia, and potentially death.39 Thus baclofen withdrawal should be considered in the differential diagnosis of an agitated child with an indwelling baclofen pump.
Major Anatomic Organization of the Nervous System
The nervous system in mammals is organized along an evolutionarily conserved axis. It can be divided anatomically and functionally into the central and peripheral nervous systems. Broadly speaking, the CNS consists of the spinal cord and the brain inside the skull. All other components, such as nerves after they leave the spinal canal or exit the brain as well as autonomic ganglia in the body, comprise the PNS. The general subdivisions are shown in Figure 57-2. The following sections focus on well-defined functions of these subdivisions as well as on their clinical relevance in pediatric critical care medicine.
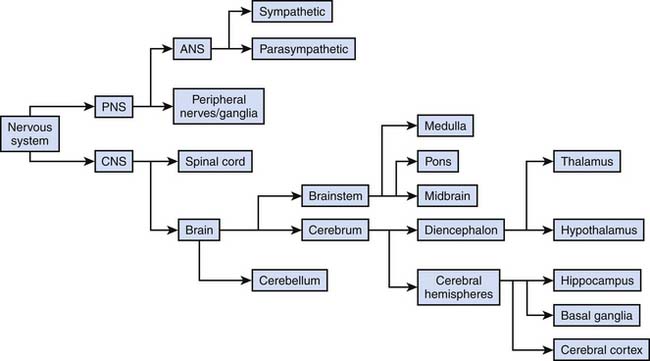
Figure 57–2 General subdivisions of the nervous system.
(Modified from Nolte J, The human brain. An introduction to its functional anatomy, ed 3, St Louis, 1993, Mosby–Year Book.)
Central Nervous System
Spinal Cord
The spinal cord is organized into segments delineated by the exiting spinal nerves. In humans, there are 31 segments: 8 cervical, 12 thoracic, 5 lumbar, 5 sacral, and 1 coccygeal.40 The first seven cervical nerves exit the spinal canal above the corresponding vertebra, that is, the C1 nerve exits between the occiput and the C1 vertebra (the atlas). Since in humans there are only seven cervical vertebra and eight cervical nerves, the last cervical nerve, C8, exits the spinal canal between C7 and T1. From T1 on, each corresponding spinal nerve exits the spinal cord below its corresponding vertebra, that is, the T12 nerve exits between T12 and L1. Early in fetal life, the spinal cord extends throughout the entire length of the spinal canal. Beginning in gestation week 12, the growth rate of the vertebrae exceeds that of the spinal cord, such that by birth in humans, the spinal cord ends at L3. During postnatal development, further differential growth occurs, and in adults, the spinal cord ends more rostrally, between L1 and L2. The nerve roots continue to exit the spinal canal through their corresponding foraminae, such that the caudal spinal roots extend past the end of the spinal cord towards their exit points and form the cauda equina. The end of the spinal cord forms an important landmark during development, since the lumbar puncture must be performed below the spinal cord in order to avoid severe injury. Hence, in infants, the preferred location of the lumbar puncture is between L4 and L5 vertebrae, with an alternate site between L3 and L4. In adults, it is safe to perform the lumbar puncture between L3 and L4, with both the L2/L3 and the L4/L5 intervertebral spaces as alternative sites.
Spinal cord lesions result in two general subsets of neurologic deficits: those caused by interruption of ascending information flow towards the brain, and those caused by interruption of descending brain control of the spinal cord circuitry and the PNS. Thus complete spinal cord transection leads to loss of sensation and muscular paralysis below the level of the lesion due to injury to the ascending sensory pathways and descending motor pathways, respectively. Immediately after the transection, paralysis is flaccid with loss of deep tendon reflexes, characteristic of spinal shock (not to be confused with neurogenic shock, see below). After a period of time, paralysis becomes spastic, with increased muscle tone and hyperactive deep tendon reflexes, due to disrupted inhibitory control of spinal cord circuitry by the brain’s motor centers.
Medulla
The medulla extends rostrally from the spinal cord in the rough shape of an ice cream scoop.40 The closed “handle” portion contains an enclosed central canal contiguous with that in the spinal cord. The open “scoop” portion is located rostrally where the central canal opens into the fourth ventricle. The medulla gives origin to four cranial nerves (CN): the glossopharyngeal (CN IX), vagus (CN X), accessory (CN XI), and hypoglossal (CN XII). It also contains decussations (crossings) of two major fiber tracts. The postsynaptic fibers from the nuclei gracilis and cuneatus, that carry tactile information from the lower and upper parts of the body, respectively, cross the midline in the caudal medulla, giving rise to the sensory decussation. The pyramidal tracts, that contain fibers descending from the motor cortex into the spinal cord, decussate slightly more rostrally, giving rise to the pyramidal, or motor decussation. These decussations are responsible for the fact that the left half of the brain controls and senses the right half of the body, and vice versa. Thus in general, damage to brain structures above the decussation gives rise to contralateral symptoms whereas damage below the decussation results in ipsilateral symptoms (except for the cerebellum as discussed below). Finally, the medulla contains the brain’s respiratory control center, which is of paramount importance in determining brain death (see below). In the absence of neuromuscular blockade, complete apnea in response to rising PaCO2 results only when the medulla has been extensively injured.
Although extensive damage to the medullary structures is often quickly fatal due to ensuing apnea, more localized injury produces a number of recognizable syndromes. Wallenberg, or lateral medullary, syndrome occurs when the territory supplied by posterior inferior cerebellar artery has been compromised, and consists of loss of temperature and pain sensation on the contralateral side of the body and on the ipsilateral side of the face. Additionally, Horner syndrome and varying degrees of vertigo, dysphagia, and dysarthria can be present. The medial medullary syndrome (Dejerine or inferior alternating syndrome) results from injury to the territory supplied by the anterior spinal artery or, occasionally, the vertebral artery. It is characterized by weakness or complete hemiplegia and loss of tactile and vibratory perception on the contralateral side of the body, together with preservation of temperature and pain sensation in the body and full sensation in the face. Medullary injury also occurs as a life-threatening, albeit rare, complication of tonsillectomy and adenoidectomy.41






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


