Chapter 60 Status Epilepticus
Definition
The International League Against Epilepsy defines SE as a seizure that persists for a sufficient length of time or is repeated enough to produce a fixed and enduring epileptic condition.1 From the practical perspective of the treating physician, this definition is difficult to apply. A more commonly used clinical definition includes duration of seizure activity, specifying SE as seizures lasting 30 minutes or longer, which is based on the theory that neuronal damage may begin after 30 minutes of continuous seizures. No clinical data support the 30-minute rule, however, and more recent definitions of SE include a shorter duration of seizure activity. An operational definition has been proposed, in which generalized convulsive SE in adults and children older than 5 years is “greater than or equal to 5 minutes of continuous seizures or two or more discrete seizures between which there is incomplete recovery of consciousness.”2 This definition has been supported by clinical studies showing that most secondarily generalized convulsive seizures in adults last less than 5 minutes3; these results suggest that seizures lasting more than 5 minutes should be regarded as more significant. Similar data in children have shown that new-onset seizures cluster by duration; most seizures (76%) last less than 5 minutes and 24% last longer, with a mean duration of 31 minutes.4 The shorter duration definition of SE remains controversial, however, because although it is less common for seizures to persist beyond 5 minutes, it also has been shown that seizures lasting 10 to 29 minutes that stop spontaneously are associated with a significantly lower mortality rate than seizures lasting beyond 30 minutes.5 Thus before the definition of SE includes a seizure duration of less than 30 minutes, further study and discussion are required.
Classification of Status Epilepticus
SE should be classified with the same terminology used for seizure classification. The International League Against Epilepsy Task Force on Classification and Terminology identifies two major categories that may be applied to both self-limited and continuous seizures.6 Generalized seizures refer to seizures involving both cerebral hemispheres, which are believed to originate within, and rapidly engage, bilaterally distributed neuronal networks. Consciousness is usually impaired. Generalized seizures include seizures with motor movements as well as absence seizures during which no convulsive signs may be present. Electroencephalography (EEG) indicates seizure activity involving both cerebral hemispheres. Focal seizures, previously referred to as partial seizures, are those in which the first clinical and EEG changes indicate seizures that originate within one cerebral hemisphere. Generalized seizures that begin as focal seizures are termed secondarily generalized seizures. Complex focal seizures are associated with an impairment of consciousness. Simple focal seizures have no impairment of consciousness. When simple focal seizures are prolonged and have a focal motor component, the term epilepsia partialis continua is used.7 These same terms are also used to describe continuous seizures and SE. SE also may be defined more specifically by seizure type, including myoclonic status, tonic status, and absence status.6 Although these distinctions are helpful to predict the cause and outcomes of SE, the differentiation may require EEG and has limited utility in the acute clinical situation.
A more clinically useful classification scheme involves categorizing SE by the presence of motor movements (Box 60-1). Convulsive SE (CSE) consists of continuous clonic or tonic motor activity with bilateral epileptiform discharges on the EEG. As CSE progresses, the motor movements are reduced and may be characterized only by subtle twitching of the limbs, face, or trunk. This condition is termed subtle CSE.
Epidemiology of Status Epilepticus
Incidence
Two population-based studies have examined the incidence of SE in the United States. The incidence of SE in Richmond, Va.,8 was determined to be 41 per 100,000 population, and in Rochester, Minn.,9 the incidence was 18 per 100,000 population. The difference may reflect a different racial composition in the communities studied; in the Richmond study, the incidence of SE was higher in the nonwhite population.
A population-based United Kingdom study determined the incidence of CSE in children to be 17 to 23 per 100,000 per year. The incidence of CSE was highest in children younger than 1 year (51 per 100,000 per year) falling to only 2 per 100,00 per year in 10- to 15-year-olds. The incidence of CSE was similar in girls and boys.10 Lower socioeconomic status, independent of ethnicity, was associated with prolonged febrile seizures and acute symptomatic CSE.11 SE was found to be the admission diagnosis of 1.6% of children admitted to a pediatric intensive care unit.12
Cause
One of the largest series of SE in children was published in 1970. This review of 239 cases of SE in children identified 47% of cases to be symptomatic (26% acute symptomatic and 21% remote symptomatic) and the remaining 53% of cases to be idiopathic, including febrile SE.13 The cause of SE is age dependent; infection plays a larger role in the cause of SE in children than in adults. In a 1996 study in Richmond, Va., the three major causes of SE in children were infection with fever, remote symptomatic cause, and low anticonvulsant drug levels (Table 60-1).8 The three major causes in adults were low antiepileptic drug (AED) levels, remote symptomatic cause, and stroke.8 The cause also differs among younger and older children. Febrile and acute symptomatic SE are more common in children younger than 2 years, whereas idiopathic and remote symptomatic causes are most common in children older than 4 years.12,14
Table 60–1 Causes of SE in Children from the Richmond, Va., SE Database
| Cause | % of SE |
|---|---|
| Non-CNS infection | 52 |
| Remote symptomatic | 38 |
| Low AED level | 21 |
| Cerebrovascular disease | 10 |
| Metabolic | 7 |
| Hypoxia | 5 |
| Idiopathic | 5 |
| CNS infection | 2 |
| Drug overdose | 2 |
AED, Antiepileptic drug; CNS, central nervous system; SE, status epilepticus.
From DeLorenzo RJ, Hauser WA, Towne AR, et al: A prospective, population based epidemiologic study of status epilepticus in Richmond, Virginia, Neurology 46:1029, 1996.
A recent prospective population-based study found that more than half of children with a first event of CSE were previously neurologically healthy. One third were identified as having prolonged febrile seizures, 17% as having acute symptomatic seizures, and 16% as having remote symptomatic seizures, and the remaining 12% had cryptogenic or idiopathic epilepsy.10 A retrospective case series of SE in children report similar results.15 Prolonged febrile seizures represent a significant burden in the PICU. A report of 119 prolonged febrile seizures, defined as lasting longer than 30 minutes, found that the seizures lasted a median of 68 minutes, and 24% lasted more than 2 hours. Analysis suggested that the longer a seizure lasts, the less likely it is to spontaneously remit.16
In children with a first seizure, 12% present in SE.17 In a report of 613 children with newly diagnosed epilepsy, 9.1% had provoked or unprovoked SE before the diagnosis of epilepsy and a nearly equal number, 9.5%, had an episode of SE following the diagnosis of epilepsy (8-year median follow-up).18,19 Children who had SE before a diagnosis of epilepsy were most likely to have remote symptomatic epilepsy. Younger age of epilepsy onset and symptomatic epilepsy increased the risk of SE.19
Outcome
Mortality
Status epilepticus is a clinical expression of potentially severe brain disease and may threaten life through impairment of airway control and respiratory function. Once intubation and mechanical ventilation are established, the outcome is primarily related to the underlying brain disease and the ability to control seizures. Age and etiology are strong predictors of outcome in SE, reflecting the fact that morbidity and mortality are more related to the underlying disorder than to the seizures. Mortality rates as high as 23% have been reported in SE in adults20,21; however, such high rates have not been reported in children. Case fatality was 3% in a prospective population-based study of first episodes of CSE in children. Death was associated with bacterial meningitis, underlying metabolic disease, and progressive neurodegenerative disorders.10 In a study of 193 children with SE, 3.6% died within 3 months of the SE; the etiology of SE was the strongest predictor of death. All deaths that occurred were associated with an acute CNS insult or progressive encephalopathy, and no children with unprovoked or febrile SE died.22 Similar findings were documented in a study of 40 cases of pediatric SE, in which mortality rate was 10% and the cause of SE in all children who died was acutely life-threatening.23 Refractory SE carries higher risks of significant morbidity and mortality (see the section on Refractory SE later in this chapter).
Subsequent Development of Epilepsy
Epilepsy, which is defined as recurrent unprovoked seizures, is a potential long-term complication of SE. In children, the risk that epilepsy will develop after SE ranges from 26% to 36%.23,24 This risk is not significantly different from rates of epilepsy following a brief first unprovoked seizure.25 As with mortality, the development of epilepsy after SE is strongly related to the etiology of SE. In children with acute symptomatic SE, the risk for development of subsequent epilepsy was found to be 41%, three times higher than that for children with self-limited acute symptomatic seizures. This risk was highest in children with anoxic encephalopathy, followed by structural and then metabolic causes.26
In contrast, children with a first unprovoked seizure presenting as SE were not more likely than children with self-limited first unprovoked seizures to have recurrent seizures.17 Furthermore, the increased risk of the development of epilepsy after febrile SE is generally reported to be small to none.22–2427
Pathophysiology
The exact mechanism that permits a seizure to become sustained is not known. Theoretically, either a failure of physiologic neuronal inhibition or an excess of excitation is required. Much of the research in this area has focused on the failure of neuronal inhibition by the major inhibitory neurotransmitter in the CNS, γ-aminobutyric acid (GABA). Prolonged seizures have been associated with deficits in GABA-mediated neuronal inhibition because of the rapid internalization of synaptic GABAA receptors. Clinically this phenomenon may be demonstrated by the poor response of prolonged seizures to the GABA agonists benzodiazepines and barbiturates,28 a phenomenon known as time-dependent pharmacoresistance.29–31
SE causes neuronal injury by both hypoxic-ischemic and excitotoxic mechanisms. The pathophysiology of SE-induced hypoxic-ischemic injury is illustrated in Figure 60-1.32 Early in SE, an increase in neuronal metabolic demand occurs with a compensatory increase in cerebral blood flow (CBF) and brain oxygenation. Later in SE, these homeostatic mechanisms are unable to keep up with the sustained increase in cerebral metabolic demand. Autoregulation of CBF fails, and consequently a higher mean arterial pressure is required to maintain adequate brain perfusion. As systemic blood pressure falls, a subsequent reduction in brain parenchymal oxygenation occurs. In the face of increased neuronal metabolic demand, uncontrolled muscular contractions, hyperthermia, and impaired airway control, both impaired oxygenation and ventilation contribute to the mismatch between metabolic supply and demand. However, neuropathologic injury occurs even when these systemic factors are controlled and before CBF is reduced, presumably because of unmet neuronal metabolic demands alone.
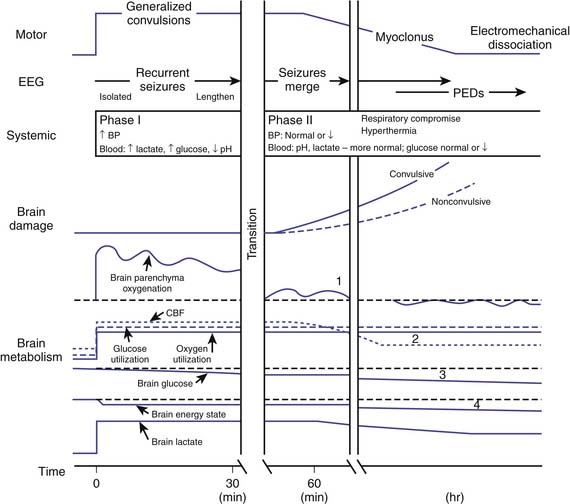
Figure 60–1 Pathophysiology of hypoxic-ischemic brain injury in status epilepticus.
(Modified from Lothman E: The biochemical basis and pathophysiology of status epilepticus, Neurology 40(5 suppl 2):13-23, 1990.)
Excitotoxicity is the second major mechanism for neuronal injury in SE. Glutamate is the major excitatory neurotransmitter in the CNS, and glutamate agonists are used to induce seizures in experimental animal models of SE. Studies have implicated both the N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) glutamate receptors in the pathophysiology of SE.33 The NMDA receptor may have a dual function in the pathophysiology of SE. Magnesium blocks the NMDA channel when it is in its normal state. With depolarization, magnesium no longer blocks the channel, and calcium, which is allowed to flow into the cell, produces further depolarization. Thus the NMDA receptor is activated when the cell is depolarized and responds by further depolarizing the cell, sustaining the excitation. This process also has been implicated in SE-induced neuronal injury, as the accumulation of intracellular calcium activates pathologic processes resulting in both necrotic and apoptotic cell death.34,35
Diagnosis
The clinical presentation of SE is variable, depending on seizure type and the baseline developmental and medical status of the child. Diagnosis depends on the identification of continuous or repetitive seizures, which is usually straightforward in the presence of convulsive seizures. However, after prolonged convulsive seizures the motor manifestations often diminish, and the seizures may become subtle or even nonconvulsive. Differentiating between such nonconvulsive seizures and a postictal state can be challenging. Patients with nonconvulsive SE, including absence SE, may be difficult to identify by history and physical examination alone. These patients can have intermittent altered awareness and continuous lethargy or unresponsiveness with or without subtle myoclonic jerks of the face or limbs. In such cases, continuous EEG monitoring is critical for diagnosis. EEG monitoring also can be used to identify nonepileptic (psychogenic) SE. Although uncommon, prolonged delays in the EEG-based diagnosis of nonepileptic SE can lead to unnecessary medical intervention.36
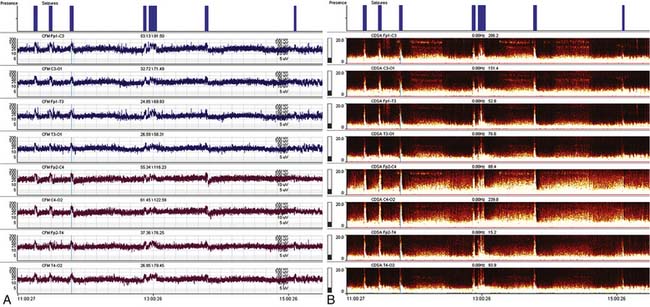
Recent advances in computing and networking technology have facilitated the remote interpretation of continuous EEG recordings. Regular communication between ICU staff and the neurology team remains essential to optimizing the care of SE patients. Quantitative EEG trending tools such as amplitude-integrated EEG and color density spectral array are increasingly being incorporated into portable EEG systems and bedside monitors (Figure 60-2). These tools may be applied to visualize single or multiple channels of EEG, providing a time-compressed overview of cerebral function that is simpler to interpret than conventional EEG. However, because these displays are generally less sensitive and specific for the identification of seizures, they should be used judiciously, with corroboration by careful analysis of the underlying raw EEG tracing.
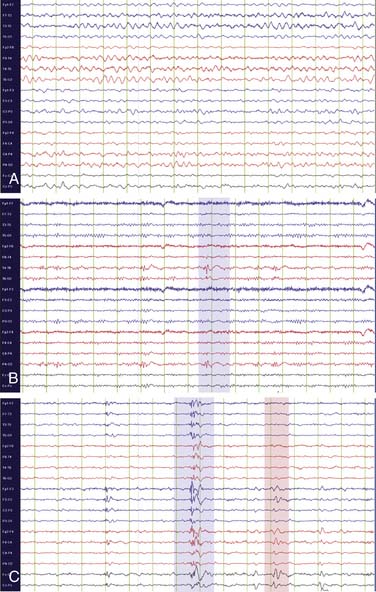
Abnormal waveforms on EEG can be divided into two categories: epileptiform abnormalities and nonepileptiform abnormalities. Epileptiform abnormalities are abnormal discharges associated with an increased risk of seizures, including sharp waves, spikes, polyspikes, and spike and slow wave discharges (Figure 60-3). When present between seizures, these waveforms are termed “interictal” abnormalities. Seizures are defined electrographically by the presence of “ictal” EEG abnormalities, whose hallmark is their rhythmicity and their evolution in frequency or distribution over the course of a seizure. Generalized seizures are characterized by widespread bilateral rhythmic epileptiform discharges (Figure 60-4, A). Focal seizures are characterized by rhythmic epileptiform discharges that are confined to one brain region (Figure 60-4, B). Focal seizures may spread to involve both cerebral hemispheres, a process termed secondary generalization.
< div class='tao-gold-member'>
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


