Chapter 61 Severe Traumatic Brain Injury in Infants and Children
The topic of severe traumatic brain injury (TBI) in infants and children has been the focus of many chapters and reviews, but it has been the focus of few clinical reports and even fewer randomized controlled trials (RCTs). The paucity of clinical trials in infants and children with severe TBI became apparent when a group of physicians and scientists (pediatric critical care medicine specialists, pediatric neurologic surgeons, pediatric emergency medicine specialists, and experts in the methodology of evidence-based medicine) met and published an evidence-based document outlining “Guidelines for the Management of Severe TBI in Infants, Children, and Adolescents” (henceforth referred to as “the pediatric guidelines” in this chapter).1 While the document focused on evidence from pediatric studies, small controlled studies or even uncontrolled case series of specific therapies for severe pediatric TBI were scarce. Studies focused on the age-related effects of therapies in the pediatric population were largely absent, and therapeutic trials in key pediatric subgroups, such as victims of inflicted childhood neurotrauma (child abuse), were rare. Based on the existing literature, few recommendations at the guideline level could be made. Subsequent to these guidelines and the last edition of this textbook, a number of studies have contributed to improving evidence-based care of children with severe TBI, and this information is incorporated into this chapter. This chapter addresses a practical and contemporary approach to the management of patients with TBI based on several sources of information: (1) the pediatric data presented in the pediatric guidelines, (2) data from studies in adults with severe TBI, and (3) accepted principles of the physiology and pathophysiology of the cerebral circulation and cranial vault.
This chapter focuses on severe TBI, specifically on management in the pediatric intensive care unit (PICU). The evolution of PICU management has progressed from exclusively supportive care to strategies attempting to (1) optimize substrate delivery and cerebral metabolism, (2) minimize secondary insults that might worsen secondary damage and outcome, (3) prevent severe or intractable intracranial hypertension and/or herniation, and (4) target specific mechanisms involved in the evolution of secondary injury with novel therapies. The goal of contemporary neurointensive care is the prevention and/or amelioration of secondary injury. The role of newer technologies and the differences between adults and children are highlighted. For information on mild or moderate TBI, outcomes, or rehabilitation of pediatric patients with TBI, the reader is referred to other materials on these topics.2,3
Epidemiology
Traumatic brain injury remains a significant pediatric health problem, with an estimated incidence of approximately 230 cases per 100,000.4 About 475,000 new cases of pediatric TBI are reported each year,5 which corresponds to a frequency of approximately one case every 2 to 3 minutes.6 TBI is the most important cause of death and disability in children.6,7 Specifically, 3000 to 4000 pediatric deaths resulting from TBI are reported annually.8 About 10% to 15% of TBI cases in infants and children are severe (Glasgow Coma Scale [GCS] score <8), and these cases contribute most of the deaths or permanent brain damage observed.9 The incidence of pediatric TBI is distributed evenly within three age groups (0 to 4 years, 5 to 10 years, and 11 to 15 years) based on data from the Traumatic Coma Data Bank.10 Although children 5 to 15 years of age generally have favorable outcomes compared with adults, children aged 4 years or younger—particularly those younger than 2 years—have a worse outcome than older children and adults. Inflicted childhood neurotrauma is the leading cause of severe TBI in infants and is believed to be the key contributor to poor outcome in this younger subgroup, although other factors may play a role.10,11 Penetrating injuries such as gunshot wounds, although not as common as either motor vehicle accidents or child abuse, also contribute significant morbidity and mortality in the pediatric population.12
Pathophysiology
Severe TBI involves a primary injury that includes direct and immediate disruption of brain parenchyma. However, not all of the effects of the primary injury are immediately apparent because damage also evolves over time and results from a cascade of biochemical, cellular, and molecular events involved in the evolution of the injury. In this chapter we define only the key factors involved. For detailed information on this topic, the reader is referred to other reviews.13–15 Secondary injuries also result from extracerebral and intracerebral insults (e.g., hypotension, hypoxemia, ischemia, and refractory intracranial hypertension) at the injury scene and in the PICU. Three basic categories of secondary mechanisms can be defined (Figure 61-1): those associated with (1) ischemia, excitotoxicity, energy failure, and resultant cell death cascades; (2) secondary cerebral swelling; and (3) axonal injury. A constellation of mediators of secondary damage and repair are involved within each category. The contribution of each mediator to outcome and the interplay among them remain poorly defined. The biochemical and molecular responses to severe TBI resulting from child abuse often are unique and generally severe.14
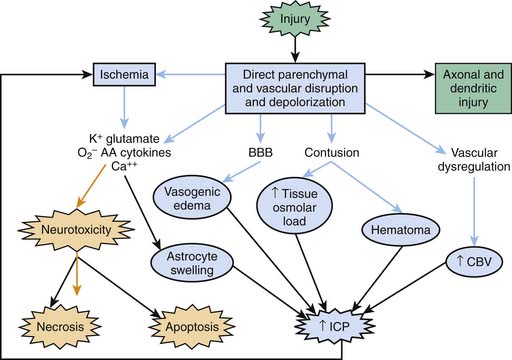
Posttraumatic Ischemia
The early studies of Pickels15 and Bruce et al.16 on cerebral blood flow (CBF) in pediatric TBI focused on the role of hyperemia in secondary brain swelling. However, clinical studies in adults showed that CBF is reduced early after injury and suggested that early posttraumatic ischemia might be of special importance.17,18 The devastating consequences of secondary extracerebral insults (i.e., hypotension and hypoxemia) early after TBI are consistent with this possibility, that is, contributing to an already compromised system. Adelson et al.19 assessed CBF in 30 infants and children after severe TBI. Early posttraumatic hypoperfusion was common, and a global CBF less than 20 mL/100 g/min was associated with a poor outcome. After the initial 24 hours, CBF often recovered, in some cases to high levels. However, delayed increases in CBF were not associated with poor outcome. This finding shifted the emphasis toward the recognition and possible treatment of hypoperfusion early after TBI to avert secondary damage.
Numerous mechanisms may contribute to early posttraumatic hypoperfusion, including (1) an attenuated vasodilatory response to nitric oxide (NO), cyclic guanosine monophosphate, cyclic adenosine monophosphate, and/or prostanoids; (2) loss of endothelial NO production; (3) elaboration of endothelin-114; and (4) production of other vasoconstrictor mediators.20 Early after injury, increases in metabolic demands resulting from uptake of glutamate are reported.21,22 Thus reduced metabolic demands coupled with CBF reduction in severely injured brain regions are an unlikely explanation for hypoperfusion. Work by Vavilala et al.11 that was published after the pediatric guidelines identified a potentially critical factor, that is, age at the time of injury, that could increase the importance of ischemic brain injury in pediatric TBI, particularly in infants and young children. These investigators reported that no difference exists in the lower limit of blood pressure autoregulation of CBF in children younger than versus older than 2 years.11 This finding led them to conclude that the autoregulatory reserve (i.e., the difference between baseline mean arterial blood pressure [MAP] and the lower limit of autoregulation) is smaller in infants than in older children or adults; even modest MAP reductions may exacerbate ischemia in infants. The contribution of this finding to secondary ischemia in pediatric TBI will become apparent later in this chapter.
Excitotoxicity
Excitotoxicity is the process by which glutamate and other excitatory amino acids cause neuronal damage. Glutamate is the most abundant neurotransmitter in the brain, but exposure to toxic levels produces neuronal death.23 Glutamate exposure produces neuronal injury in two phases. Sodium-dependent neuronal swelling quickly occurs,24 followed by delayed, calcium-dependent degeneration. These effects are mediated through both ionophore-linked receptors, labeled according to specific agonists (N-methyl-D-aspartate [NMDA], kainite, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid), and receptors linked to second messenger systems (i.e., metabotropic receptors). Activation of these receptors leads to calcium accumulation via receptor-gated or voltage-gated channels or through release of intracellular stores. Increased intracellular calcium concentration triggers a number of processes that can lead to cell death. One mechanism involves activation of constitutive NO synthase, leading to NO production, peroxynitrite formation, and resultant deoxyribonucleic acid (DNA) damage. Poly(ADP-ribose) polymerase is an enzyme operative in DNA repair. In the face of excessive DNA damage, poly(ADP-ribose) polymerase activation leads to adenosine triphosphate (ATP) depletion, metabolic failure, and necrotic cell death.25
In cerebrospinal fluid (CSF) from adults with TBI, glutamate levels were about fivefold greater than in control patients (up to 7 μm/L).26 These levels are sufficient to cause neuronal death in cell culture. Increased levels of glutamate also are seen in CSF of infants and children with TBI and correlate with poor outcome and child abuse as an injury mechanism.27 Antiexcitotoxic therapies improve outcome after experimental TBI; pretreatment with NMDA antagonists (e.g., MK-801) attenuate behavioral deficits.28 Other therapies that modify glutamate-NMDA receptor interaction and improve outcome after experimental TBI are magnesium, glycine site antagonists, hypothermia, and pentobarbital.14 Despite this benefit, clinical trials with antiexcitotoxic therapies have been unsuccessful in adults, perhaps because of adverse effects of these drugs, delayed treatment, or the antiexcitotoxic effects of many current therapies (e.g., barbiturates, hypothermia, and sedatives).29 Developing neurons are more susceptible to excitotoxic injury than are mature cells; however, concerns have been raised about use of NMDA antagonists in infants because these drugs may induce apoptotic neurodegeneration.30 Investigating which specific NMDA receptor subunits may need to be targeted to produce antiexcitotic effects without triggering apoptosis is a key area for future research.
Apoptosis Cascades
Cells that die after TBI can be categorized on a morphologic continuum ranging from necrosis to apoptosis.31,32 Apoptosis is a morphologic description of cell death defined by cell shrinkage and nuclear condensation, internucleosomal DNA fragmentation, and formation of apoptotic bodies.33 In contrast, cells that die of necrosis display cellular and nuclear swelling with dissolution of membranes (see also Chapter 100).
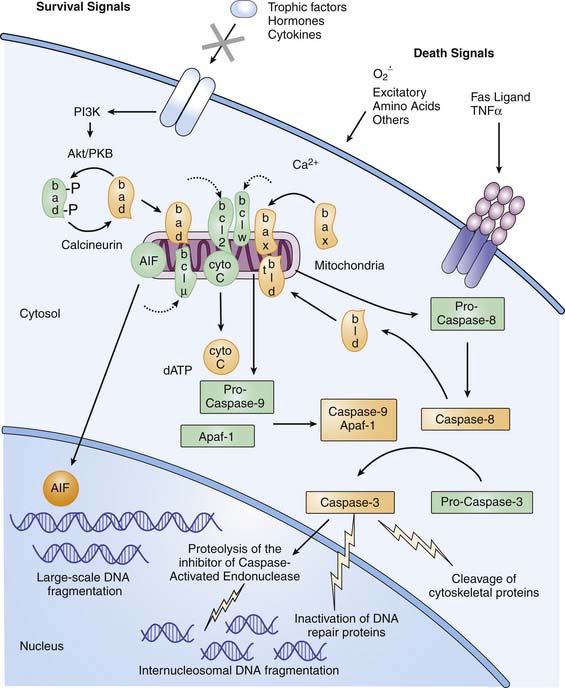
Because apoptosis requires a cascade of intracellular events for completion of cell death, “programmed cell death” is the currently accepted term for the process that leads to apoptosis. In diseases with complex and multiple mechanisms, such as TBI, distinguishing clinical apoptotic from necrotic cell death as classically defined may be difficult,34 and some cells have mixed phenotypes. In mature tissues, programmed cell death requires initiation via either intracellular or extracellular signals (Figure 61-2). Intracellular signaling appears to be initiated in mitochondria, triggered by disturbances in cellular homeostasis such as ATP depletion, oxidative stress, or calcium fluxes.35 Mitochondrial dysfunction leads to egress of cytochrome c into the cytosol. Oxidation of the mitochondrial lipid cardiolipin may play a central role in cytochrome c release.36 Cytochrome c release can be blocked by antiapoptotic members of the Bcl-2 family (e.g., Bcl-2, Bcl-xL, Bcl-w, and Mcl-1) and promoted by proapoptotic members of the Bcl-2 family (e.g., Bax, Bcl-xS, Bad, and Bid).37 Cytochrome c in the presence of dATP and a specific apoptotic-protease activating factor in cytosol activates the initiator cysteine protease caspase-9.38 Caspase-9 then activates the effector cysteine protease caspase-3, which cleaves cytoskeletal proteins, DNA repair proteins, and activators of endonucleases.39 Extracellular signaling of apoptosis occurs via the tumor necrosis factor (TNF) superfamily of cell surface death receptors, which includes TNFR-1 and Fas/Apo1/CD95.40 Receptor-ligand binding of TNFR-1–TNF-α or Fas-FasL promotes formation of a trimeric complex of TNF- or Fas-associated death domains. These death domains contain caspase recruitment domains, ultimately leading to caspase-3 activation, where the mitochondrial and cell death receptor pathways converge (see Figure 61-2). Both the intrinsic and extrinsic pathways may contribute to the evolution of cell death after severe TBI in infants and children. CSF levels of the anti-apoptotic protein Bcl-2 in pediatric patients after TBI were increased approximately fourfold in patients with TBI versus control subjects.41 Similarly, CSF levels of sFas receptor and sFas ligand are increased in patients with TBI versus control subjects.42 Apoptosis may be an important therapeutic target for new therapies in infants with severe TBI. Finally, current therapies likely attenuate both necrotic and apoptotic injury cascades.
Cerebral Swelling
After the initial minutes to hours of posttraumatic hypoperfusion and hypermetabolism, a phase of metabolic depression occurs. Cerebral metabolic rate of oxygen (CMRO2) decreases to approximately one third of normal43,44 and is maintained at that level for the duration of coma, unless perturbed by second insults such as seizures or spreading depression.45 The exact etiology of this state remains to be defined; however, contributions from reduced synaptic activity and mitochondrial failure may be important.46 Sustained increases in glycolysis are reported in some cases, possibly related to seizure activity or sustained increases in glutamate levels.22 During the PICU phase, cerebral swelling develops and generally peaks between 24 and 72 hours after injury, although sustained increases in intracranial pressure (ICP) for 1 week or longer occasionally are observed.
Cerebral Blood Volume
Several mechanisms may contribute to intracranial hypertension in infants and children after severe TBI (see Figure 61-1). Brain swelling and accompanying intracranial hypertension contribute to secondary damage in two ways. Intracranial hypertension can compromise cerebral perfusion leading to secondary ischemia, and it can produce the devastating consequences of deformation through herniation syndromes. Bruce et al.16 described the phenomenon of “malignant posttraumatic cerebral swelling” in children. CBF was measured in six children, and hyperemia was believed to be the major culprit. Muizelaar et al.,47 in a series of 32 children, and Beyda48 suggested similar findings. However, Sharples et al49,50 suggested that hyperemia was uncommon after severe TBI in children; rather, reduced CMRO2 was associated with poor outcome.
Suzuki51 measured CBF in 80 normal children. He showed an age dependence of CBF, with rather high values in children ages 2 to 9 years, levels previously suggested to represent posttraumatic hyperemia. Nevertheless, in patients with TBI, after resolution of the aforementioned early posttraumatic hypoperfusion, CBF may increase to levels greater than metabolic demands in some children, producing a state of relative hyperemia.14
Bergsneider et al.22 posed the alternative hypothesis of “hyperglycolysis” to explain the increases in CBF in patients with severe TBI whose CBF is uncoupled from CMRO2. Cerebral glutamate uptake is coupled to glucose utilization by glycolysis in astrocytes. Recent studies suggest two other potential contributors to increased glycolysis after TBI even in the absence of low CBF, namely mitochondrial falure52 and nitration and inactivation of the enzyme pyruvate dehydrogenase, which is critical to the production of acetyl-CoA and thus oxidative metabolism.53 Thus in injured brain regions with reduced CMRO2, increases in CBF may be coupled to local increases in glucose utilization even in the absence of ischemia.
Local or global increases in glycolysis occur in adults with severe TBI.22 The incidence and/or importance of secondary “hyperemia” or hyperglycolysis in pediatric TBI remains to be determined. It may occur in select cases, but secondary increases in CBF probably are not the major contributor to raised ICP. Increases in CBF were not associated with raised ICP in adults,54 and hyperemia was not associated with poor outcome in children.4 The contribution of hyperemia (increased cerebral blood volume [CBV]) to the development of raised ICP has been studied in adults with TBI.55 Increased CBV was seen in only a small number of patients. These studies suggest that the importance of posttraumatic hyperemia may have been overstated, and edema rather than hyperemia may be the predominant contributor to brain swelling after TBI.56
Edema
Both cytotoxic and vasogenic edema may play important roles in cerebral swelling (Figure 61-3). However, the traditional concept of cytotoxic and vasogenic edema is evolving. There appear to be four mechanisms for edema formation in the injured brain. First, vasogenic edema may form in the extracellular space as a result of blood-brain barrier (BBB) disruption. Second, cellular swelling can be produced in two ways. Astrocyte swelling can occur as part of the homeostatic uptake of substances such as glutamate. Glutamate uptake is coupled to glucose utilization via a sodium/potassium adenosine triphosphatase, with sodium and water accumulation in astrocytes. Swelling of both neurons and other cells in the neuropil can result from ischemia- or trauma-induced ionic pump failure. Finally, osmolar swelling may contribute to edema formation in the extracellular space, particularly in contusions. Osmolar swelling actually is dependent on an intact BBB or an alternative solute barrier. Cellular swelling may be of greatest importance.
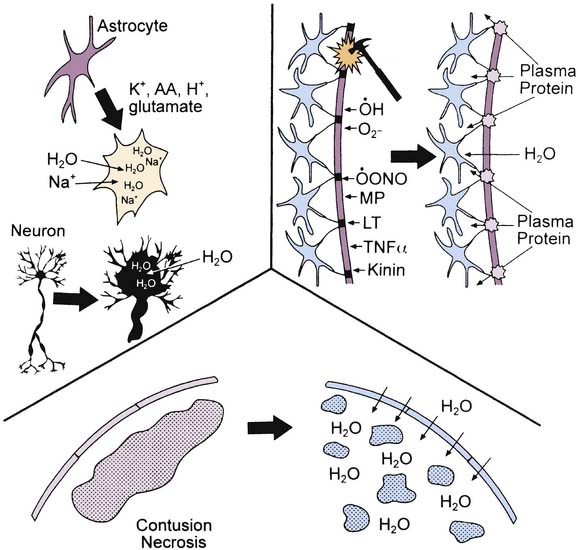
Using a model of diffuse TBI in rats, Barzo et al.57 applied diffusion-weighted magnetic resonance imaging to localize the increase in brain water. A decrease in the apparent diffuse coefficient after injury suggested largely cellular swelling rather than vasogenic edema in the development of raised ICP. Katayama et al.58 also suggested that the role of BBB in the development of posttraumatic edema may have been overstated, even in the setting of cerebral contusion. An intriguing possibility is that as macromolecules are degraded within injured brain regions, the osmolar load in the contused tissue increases. As the BBB reconstitutes (or as other osmolar barriers form), a large osmolar driving force for local accumulation of water develops, resulting in the marked swelling so often seen in and around cerebral contusions. Thus in either diffuse injury or focal contusion, BBB permeability may play only a limited role in the development of cerebral swelling. If these results can be generalized, then hypertonic saline solution or mannitol seem to represent optimal therapies. This suggestion is in contrast to traditional recommendations, in which hyperventilation is suggested.59,60 However, a role for BBB permeability in cases of severe TBI should not be dismissed. A recent study in adults with severe TBI by Polderman et al.61 reported that prolonged use (>48 hours) of mannitol (a large molecule that does not cross the intact BBB) was associated with a progressive accumulation of mannitol in CSF, and in some cases, a reverse osmotic gradient was even established. This finding suggests that breaching of the BBB is important and that prolonged use of mannitol might create an iatrogenic secondary insult. Studies of the extent of BBB injury and the contribution of cellular swelling to intracranial hypertension in pediatric TBI are needed.
Axonal Injury
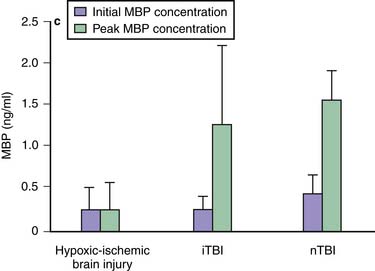
Traumatic axonal injury (TAI) encompasses the spectrum from mild to severe TBI.62,63 The extent and distribution of TAI depend on injury severity and category (focal vs. diffuse). Its incidence and nature appear to be age independent,64 but its consequences may be particularly devastating in children.65 The effects of TAI in children during a period of developmental axonal connectivity remain unknown but likely are considerable. During development, numerous signaling molecules can function as attractants or repellents.66 Clinical data on TAI after pediatric TBI are limited. However, strongly supporting the role for TAI in pediatric TBI, after publication of the guidelines, Berger et al.67 reported that serum levels of myelin basic protein are markedly increased in infants and children after either accidental or inflicted TBI—in contrast to hypoxic-ischemic encephalopathy (Figure 61-4). In children affected by shaken baby syndrome, TAI may be highly prevalent.68 The classic view that TAI occurs because of immediate physical shearing is represented primarily in patients with severe injury in whom frank axonal tears occur. Experimental studies suggest that TAI occurs by a delayed process termed secondary axotomy, which results from either calcium accumulation or altered axoplasmic flow.69 What remains to be determined is how much of TAI results from a reversible evolution of damage to axons versus Wallerian degeneration of disconnected axons. The former but not the latter would be amenable to treatment. Nevertheless, TAI contributes to the morbidity after TBI.69 Laboratory studies suggest that hypothermia, calpain antagonists, and cyclosporine A can attenuate TAI, but clinical data are lacking.
History
Unlike in adult TBI, where the history is generally straightforward, the special case of inflicted childhood neurotrauma contributes to increased importance of the history in pediatric TBI. For a discussion of the topic, the reader is referred to Duhaime70 (see also Chapter 113). In cases of severe TBI resulting from child abuse, a history that is incompatible with the observed injury is often given.71 Occult presentations of inflicted childhood neurotrauma can be particularly important because they may be recognized as cases of severe TBI relatively late in their treatment course.72 In this setting, brain edema already may have evolved to life-threatening levels, and other superimposed secondary insults (e.g., seizures and apnea) may complicate management and worsen outcome.
Signs and Symptoms
The GCS score73 (Table 61-1), first described in 1974, remains a valuable tool for grading and communicating severity of neurologic injury after TBI, although limitations remain with pediatric use. The verbal and motor components of the GCS score have been modified for assessment of infants,74 but this modification has not been validated in infants and young children. The motor score has probably become the most important component of the GCS. A rapid “mini-neuroassessment” that allows evaluation of the patient’s level of consciousness, pupillary size and light response, the fundi, extraocular movements, response of extremities to pain, deep tendon reflexes, and brainstem reflexes should all be part of the initial evaluation.75 Until proven otherwise, an altered level of consciousness, pupillary dysfunction, and lateralizing extremity weakness in an infant or child should raise suspicion of a mass lesion that may require surgery.76 These signs of impending herniation require an immediate response, as outlined in Figure 61-5.
| Glasgow Coma Scale | Modified Coma Scale | Point Scale |
|---|---|---|
| EYE OPENING | ||
| Spontaneous | Spontaneous | 4 |
| To speech | To speech | 3 |
| To pain | To pain | 2 |
| None | None | 1 |
| VERBAL | ||
| Oriented | Coos, babbles | 5 |
| Confused | Irritable | 4 |
| Inappropriate words | Cries to pain | 3 |
| Grunting | Moans to pain | 2 |
| None | None | 1 |
| MOTOR | ||
| Follows commands | Normal spontaneous movements | 6 |
| Localizes pain | Withdraws to touch | 5 |
| Withdraws to pain | Withdraws to pain | 4 |
| Abnormal flexion | Abnormal flexion | 3 |
| Abnormal extension | Abnormal extension | 2 |
| Flaccid | Flaccid | 1 |
Initial Resuscitation
The identification and correction of airway obstruction, inadequate ventilation, and shock take priority over a detailed neurologic assessment.77 Thus the first step in managing a patient with a head injury patient is complete, rapid physiologic resuscitation.1 Although raised ICP and cerebral herniation are the major complications, brain-specific interventions in the absence of signs of herniation or other neurologic deterioration currently are not recommended. Mannitol may be counterproductive for the management of malignant intracranial hypertension during initial resuscitative efforts. In the acute TBI resuscitation setting, hypertonic saline solution may be a better alternative (discussed later in this chapter). Studies have consistently shown that increased morbidity and mortality are associated with the secondary insults of hypotension and hypoxemia.78,79 Gentleman79 reported that the increased use of tracheal intubation and ventilation produced a concomitant reduction in episodes of hypoxemia and mortality rate and an increase in favorable outcomes. Although the basis for this improvement may be multifactorial, early correction of hypoxemia and hypovolemia must be the initial objective. However, specific recommendations for intubation at the scene are complex and likely are influenced by the expertise of caregivers in the field and by the transport distance, among other factors.1
We previously outlined the use of the mnemonic SOAP to define the components for optimal preparation for securing the airway in the case of severe TBI requiring intubation in the emergency department or the PICU.80 “S” represents suction; for most patients, a flexible 10 Fr catheter suffices. However, for school-age children (older than 5 years), we recommend the Yankauer rigid plastic suction catheter, which allows direct oropharyngeal suctioning. “O” represents oxygen; oxygen (a fraction of inspired oxygen [FIO2] of 1.0) should be delivered to the patient by face mask immediately before intubation. Optimal positioning of the patient requires immobilization of the neck to stabilize the cervical spine. Delivery of 100% oxygen facilitates nitrogen washout from the functional residual capacity, maximizing alveolar oxygenation. “A” represents airway; once the patient is positioned and ventilation and oxygenation are controlled, an age-appropriate laryngoscope blade and tracheal tube are selected. The tube is secured with adhesive tape that should not pass circumferentially around the neck because cerebral venous return may be reduced. “P” represents pharmacology; the medications chosen must be potent and rapid in their onset of action. The goals of analgesia, amnesia, and neuromuscular blockade must be met rapidly. Ideally, the patient never receives a preintubation positive-pressure breath.
Tracheal intubation of the child with severe TBI requires a cerebroprotective, rapid-sequence technique when possible (see also Chapter 42). Bag-valve-mask positive-pressure ventilation should be avoided. However, in cases of hypoxemia or impending herniation, positive-pressure ventilation should be instituted immediately.81 Because the bag-valve-mask technique may cause unintentional cervical spine manipulation, care is advised.82
If a person who has sustained TBI meets any of the criteria listed in Box 61-1, assisted ventilation is indicated.81,83 In children, the recommended route of initial airway control is orotracheal intubation under direct vision.84 Nasotracheal intubation should be avoided. Blind passage of the endotracheal tube around the acute nasopharyngeal angle makes this procedure an unnecessary obstacle to rapid, physiologic resuscitation. Orotracheal intubation can be accomplished using a two-person strategy that protects the cervical spine from injury. A normal lateral cervical spine roentgenogram is reassuring but does not rule out cervical spine injury.85 Spinal immobilization must be maintained,86 which is accomplished via in-line cervical immobilization by one operator while the second intubates the trachea. Care must be taken to avoid pressing into the soft tissues of the submental region and strap muscles because inadvertent airway obstruction may ensue.
Rapid-Sequence Induction and Intubation
Tracheal intubation, although lifesaving, is a noxious stimulus. The technique of rapid-sequence induction and intubation secures the airway of an unprepared patient, who is at risk for aspiration of gastric contents, in an immediate and safe manner. No resistance is provided to direct laryngoscopy, and the normal responses to intentional placement of a foreign body into the trachea are eliminated. Rapid-sequence induction is documented to be a safer technique than either nasotracheal intubation or orotracheal intubation without neuromuscular blockade.87,88
For a victim in cardiac arrest, cardiopulmonary resuscitation should begin immediately, accompanied by direct orotracheal intubation. No pharmacologic adjuncts are necessary to secure the airway. For a hemodynamically unstable patient, the combination of fentanyl, lidocaine, and rocuronium bromide is the first choice (Table 61-2). At Pittsburgh Children’s Hospital use of etomidate has been eliminated in children with TBI because of concerns about adrenal suppression.89 Either of these same sequences of drugs can be used in hemodynamically stable patients, for whom a rapidly acting benzodiazepine (midazolam) can be added.
Table 61–2 Drugs for Intubation of the Head-Injured Child
| Situation | Drugs |
|---|---|
| Cardiopulmonary arrest | Resuscitation drugs |
| Hemodynamically unstable | Fentanyl 2–4 μg/kg |
| Lidocaine 1 mg/kg | |
| Rocuronium 1 mg/kg or vecuronium 0.3 mg/kg | |
| Hemodynamically stable | Fentanyl 2–4 μg/kg |
| Lidocaine 1 mg/kg | |
| Midazolam 0.1–0.2 mg/kg | |
| Rocuronium 1 mg/kg or vecuronium 0.3 mg/kg | |
| or | |
| Thiopental 4–5 mg/kg | |
| Lidocaine 1 mg/kg | |
| Rocuronium 1 mg/kg or vecuronium 0.3 mg/kg |
An alternative in the hemodynamically stable patient is thiopental. Thiopental rapidly reduces cerebral metabolism,90–93 which in turn blunts the ICP rise associated with laryngoscopy. The rapid cerebral uptake of these agents is matched by rapid washout. Thus these agents must be followed with another sedative or analgesic. Fentanyl, in combination with lidocaine, reduces the catecholamine surge associated with direct laryngoscopy.81
Circulatory Stabilization
Assessment of circulatory function after trauma involves the rapid determination of heart rate, blood pressure, central and peripheral pulse quality, capillary refill, and cerebral perfusion.94 Posttraumatic hypoperfusion must be assumed to be hypovolemic (i.e., hemorrhagic) in nature, but it also may have a secondary component of myocardial depression resulting from cardiac contusion. However, cardiac contusion is less common in children than in adults. In patients with severe TBI, fluid therapy for hypovolemic shock entails rapid replacement of vascular volume (see also Chapter 29). The choice of resuscitation fluid is controversial. The current recommendation is 20 mL/kg isotonic crystalloid (0.9% NaCl solution) given as soon as vascular access is obtained. Hypotonic fluid should not be used in the initial resuscitation of a patient with a brain injury. Subsequent doses of fluid should be isotonic crystalloid, colloid, or packed red blood cells and titrated based on serial assessment of blood pressure, perfusion, and hematocrit. Although concerns exist regarding the relative hypotonicity of lactated Ringer’s solution,95 evidence from studies in laboratory animals supports the safety of the use of lactated Ringer solution in patients with TBI.96 Fisher et al.97 reported the successful use of 3% saline solution as a maintenance fluid in children with TBI. Titration of 3% saline solution as an infusion to prevent development of intracranial hypertension is an acceptable first-tier strategy. Although its efficacy has yet to be proved in a clinical trial, inclusion of hypertonic saline solution should be considered in the setting of volume resuscitation of a child with TBI who has initial signs or symptoms of intracranial hypertension. Details of the approach to osmotherapy are discussed later in this chapter.
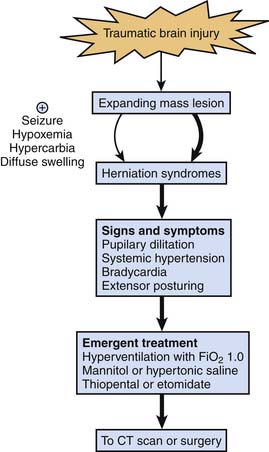
Herniation
The need to simultaneously address airway control, cardiovascular assessment and stabilization, treatment of extracerebral insults (hemorrhage and multiple trauma), and initial trauma survey in the field and emergency department makes management challenging. Although mass lesions are less common in children than in adults, they still occur in about 30% of children with severe TBI.6 Although it happens infrequently, some of these patients, particularly those with rapidly expanding mass lesions (e.g., epidural hematoma), can present with signs and symptoms of herniation (i.e., pupillary dilatation, systemic hypertension, bradycardia, and extensor posturing). Because the devastating complications of herniation sometimes can be prevented or treated in the initial minutes of their progression, the importance of aggressively and presumptively treating signs and symptoms of herniation, which is a medical emergency, cannot be overemphasized until these signs and symptoms are proved not to represent herniation.
Appropriately, there has been a move away from prophylactic application of aggressive hyperventilation for the management of severe TBI. However, it is important to recognize that temporary use of hyperventilation with an FIO2 of 1.0 is a therapy that can be immediately applied and can be life saving in the setting of impending herniation until other therapies can be instituted. Intubating doses of thiopental or pentobarbital and mannitol (0.25 to 1.0 g/kg) or hypertonic saline solution97 also should be given emergently. One must recognize that factors other than a focal mass lesion may lead to herniation and that these situations may arise more commonly in children than in adults. Diffuse swelling is more common in children than in adults, and in this setting, inadvertent hypercarbia or hypoxemia, iatrogenic excessive fluid administration, or status epilepticus can precipitate herniation. Although discussed in this chapter in the context of acute therapy, herniation can occur at any time in the PICU course, and this approach to treatment also applies (see Figure 61-5).
Transition from the Emergency Department to the Pediatric Intensive Care Unit: Computed Tomographic Scan and Intracranial Pressure Monitoring
The transition of patients with severe TBI from the emergency department to the PICU includes computed tomographic (CT) evaluation of the head (and other anatomic regions, when clinically indicated) followed by placement of an ICP monitor, transport to the operating suite for surgical intervention, or both. In the initial resuscitation, sedation must be carefully titrated, maintaining the difficult balance that produces stability, analgesia, and anxiolysis during transport and scanning while allowing for rapid emergence for clinical assessment (as indicated) until a decision is made regarding surgery or ICP monitoring. Because the early period after injury generally reflects a state of increased vulnerability of the brain to second insults because of the brain’s increased metabolic demands and compromised perfusion, providing adequate sedation and maintaining stable hemodynamics are important. An end-tidal CO2 monitor also should be considered to avoid iatrogenic hyperventilation or hypoventilation. Clinical trials supporting definitive recommendations are not available. Nevertheless, the risks of intrahospital transport are well described;98 therefore, when possible, a physician should accompany the infant or child upon transport to the scanner to direct care because serial assessment to titrate therapy is needed during the acute phase of injury.
Diagnostic Studies and Monitoring Modalities
Computed Tomography
Since becoming commercially available in 1973, CT has been of enormous benefit to neurointensive care.99 Examples of classic findings in severe pediatric TBI are shown in Figure 61-6. Comprehensive classifications of CT findings in adults with severe TBI are reported. The Marshall classification is the most commonly used (Table 61-3).100 A similar system specifically for pediatric TBI is not described, although several reviews characterize the spectrum of injury in infants and children as defined by CT.101,102 Ewing-Cobbs et al.101 compared acute CT findings in infants and children with inflicted and noninflicted injuries. Subdural interhemispheric and convexity hemorrhages and preexisting lesions were two to three times more common in the group with inflicted TBI. Epidural hematomas were more common in the group with noninflicted TBI.
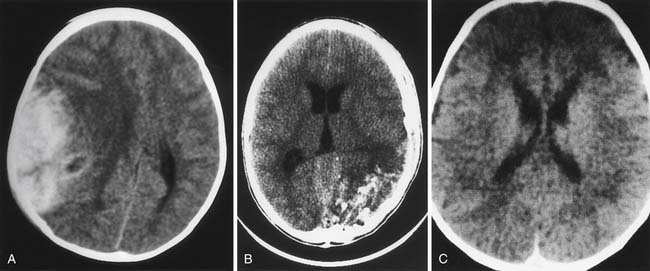
Table 61–3 Marshall Classification of Cranial Computed Tomographic Scans
| Classification | Findings on Scan |
|---|---|
| Diffuse injury I (no visible pathologic change) | No visible intracranial pathologic change seen on computed tomography |
| Diffuse injury II | Cisterns are present with shift 0–5 mm and/or lesion densities present |
| No high or mixed density lesion >25 mL | |
| May include bone fragments and foreign bodies | |
| Diffuse injury III | Cisterns compressed or absent (swelling) with shift 0–5 mm |
| No high or mixed density lesion >25 mL | |
| Diffuse injury IV (shift) | Shift >5 mm |
| No high or mixed density lesion >25 mL | |
| Evacuated mass lesion | Any surgically evacuated lesion |
| Nonevacuated mass | High or mixed density lesion >25 mL, not surgically evacuated |
| Brain dead | No brainstem reflexes |
| Flaccidity | |
| Fixed and nonreactive pupils | |
| No spontaneous respirations with a normal PaCO2 | |
| Spinal reflexes permitted |
Timing of repeat cranial CT scans has been investigated, including studies in children. Routine reimaging at 24 or 48 hours after injury has been suggested.103 However, Tabori et al.104 evaluated the impact of routine reimaging on 67 children after severe TBI and noted that although some new lesions were identified, reimaging did not lead to surgical or medical changes in therapy in any patient. A decision to reimage based on changes in ICP or clinical examination was recommended. Such an approach also is recommended in published pediatric guidelines.1
Studies in adults and children indicate that CT scans are not without limitations and must be used as only one, albeit important, piece of information. After severe TBI, in about 15% of adults with a normal CT scan, clinically significant intracranial hypertension develops. In contrast, in a study in 65 children, Hirsch et al.105 reported that CT scans had a high false-positive rate in defining increased ICP. Finally, patients with normal initial head CT scans who also have hypotension or abnormal posturing have the same propensity to the development of intracranial hypertension as do their counterparts with an abnormal scan.32,33
Magnetic Resonance Imaging
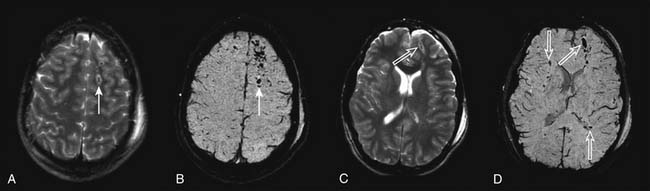
MRI may have future applications salient to acute management in persons with TBI. The application of diffusion-weighted MRI for studying the evolution of cerebral edema,55 the use of novel MRI methods for quantifying CBF,106 and new methods such as susceptibility-weighted imaging to assess white matter damage increasingly are being applied (Figure 61-7).107 The potential ability to couple these techniques with MR spectroscopy and functional MRI are beginning to yield unprecedented advances in our understanding of the brain’s response to injury. However, MRI suites in most institutions are remote from the emergency department and PICU, introducing the risk of transport. Currently, hardware incompatibilities (e.g., ventilators and intravenous pumps) and long data acquisition times (relative to CT) limit the utility of this important tool.
Intracranial Pressure Monitoring
Unfortunately, clinical signs such as pupillary size, light response, and papilledema fail as early indicators of intracranial hypertension. Although the most reliable clinical signs are those associated with herniation, the introduction of ICP monitoring devices has allowed detection of intracranial hypertension before such changes are observed.108,109 In the pediatric guidelines,1 ICP monitoring was suggested as appropriate in children with an abnormal admission head CT scan and initial GCS score between 3 and 8. Also, ICP monitoring was suggested to be appropriate in adults with severe TBI and a normal head CT scan if the clinical course was complicated by hypotension or motor posturing. This modality is essential to implementation of a physiologically guided approach to management of cerebral perfusion pressure (CPP) in the infant or child with severe TBI.110
ICP monitoring has not been studied in an RCT to establish its efficacy in altering outcome after severe TBI in either adults or children. Since publication of the pediatric guidelines, however, several studies have contributed new insight. Forsyth et al.111 recently examined the United Kingdom multicenter database of more than 500 cases of pediatric TBI and reported that both ICP >20 mm Hg and lack of ICP monitoring were independently associated with death before discharge. Given that raised ICP correlates with poor outcome, there is a strong rationale for identifying and treating this problem (see also Chapter 59).112,113 As discussed in the section on CT, although CT is useful for identifying patients at high risk for the development of raised ICP (e.g., those with mass lesions), the finding of a “normal” cranial CT scan does not rule out the potential for raised ICP.114 Despite this evidence, ICP monitoring in children with TBI, particularly in infants, is still not rigorously performed in clinical practice. Keenan et al.115 surprisingly reported in 2005 that only 33% of infants and young toddlers (younger than 2 years) with severe TBI underwent ICP monitoring in the state of North Carolina. Consideration of risk versus benefit for ICP monitoring must be involved in the clinical decision in cases in which the complication rate is high, such as in patients with coagulopathy.
Currently, ICP monitoring by ventricular catheter is considered the most accurate, low-cost, reliable method.1 The ventricular catheter also affords a key therapeutic option—CSF drainage. Other acceptable methods include parenchymal fiberoptic and microtransducer systems; subarachnoid, subdural, and epidural monitors of any type are less reliable.116 The type of monitor (ventricular catheter or fiberoptic pressure transducer) used is dependent on the local preference of the neurosurgical staff.
Monitoring and treatment of ICP are essential to contemporary management. Use of a ventricular catheter affords the added opportunity of CSF drainage as a therapy. Adult patients who have severe TBI with an ICP higher than 20 mm Hg have a poorer outcome than do those without increased ICP.116 Similarly, although a large prospective RCT of children with and without both ICP monitoring and CPP management has not been performed, a prospective cohort study suggested a better outcome in adults monitored and treated with CSF drainage compared with those who did not undergo ICP monitoring.117 However, this topic is controversial in the adult TBI literature,118 and further study is needed.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


