Chapter 123 Sedation and Analgesia
Patients may recall their stay in the ICU. Many patients remember having an endotracheal tube or having their lungs mechanically ventilated. Nightmares and hallucinations also have been reported.1 Either single-drug therapy or inadequate dosing may be associated with a higher incidence of recall in the patient receiving neuromuscular blocking agents. 2 In adults, delusional memories and an underlying anxiety state were predictors of the development of a posttraumatic stress disorder after sedation in the ICU.3 Delusional memories were reported much more frequently than factual memory, probably because most patients have difficulty correctly remembering the events that occur during their stay in the ICU. In pediatric patients, recall of the PICU experience also has been reported.4 More than 66% of pediatric patients remembered their stay in the PICU. Eighteen percent had bad memories, 16% remembered mechanical ventilation and anxiety, and 29% remembered pain from a procedure or movement. Overall the recollections of patients in the PICU were considered negative in approximately 15% of the patients. Sleep disturbance also was a problem.
Various scoring systems are often used to guide sedation. The most widely used is the Ramsay scale.5 The patient’s level of consciousness is classified as one of six scores (Table 123-1). The nurse at the bedside assesses the patient and then changes the sedation regimen as necessary to achieve the desired level of sedation. The ideal level of sedation varies from patient to patient, but in general, most intensive care physicians seek to maintain patients in a sleepy but easily awakened state. A Ramsay score of 2 to 3 seems to be ideal as the clinical end point for sedation. Deeper sedation should be reserved for select patients who are often younger, are receiving neuromuscular blocking agents, or have a head injury. The use of a sedation scoring system to guide sedation of surgical critical care patients has been evaluated for cost-effectiveness. Use of scoring systems has proved to save costs in the ICU.6 Because the patient can be weaned more rapidly from the ventilator through better control of the sedation level, the number of days a patient is connected to a ventilator is reduced. The COMFORT score, which is composed of eight variables (each with five categories), also has been validated for use in the PICU to assess sedation level in children. Use of this system, however, is more complicated and time consuming.7
| Level | Description |
|---|---|
| 1 | Patient awake, anxious, and agitated or restless or both |
| 2 | Patient awake, cooperative, oriented, and tranquil |
| 3 | Patient awake, responds to command only |
| 4 | Patient asleep, brisk response to light glabellar tap or loud auditory stimulus |
| 5 | Patient asleep, sluggish response to light glabellar tap or loud auditory stimulus |
| 6 | Patient asleep, no response to light glabellar tap or loud auditory stimulus |
Many scoring systems are subjective and are limited by interobserver variability. The more objective methods may be too cumbersome for routine use. A simple scoring system has been devised that is easy to use and minimizes subjectivity and observer variability.8 This system is the Brussels sedation scale. It is similar to the Ramsay scale, but the Brussels scale levels that correspond to the Ramsay scale levels 4 and 5 are better differentiated (Table 123-2).
Table 123–2 Brussels Sedation Scale
| Level | Description |
|---|---|
| 1 | Unable to be aroused |
| 2 | Responds to painful stimulation (trapezius muscle pinching) but not auditory stimulation |
| 3 | Responds to auditory stimulation |
| 4 | Awake and alert |
| 5 | Agitated |
The Bispectral Index (BIS) is a processed electroencephalogram (EEG) monitor that measures the hypnotic effects of anesthetics and sedatives. The BIS is an empirical, statistically derived measurement. The BIS monitor reports a single number from 0 to 100 that represents an integrated measure of cerebral electrical activity. The BIS has been validated as a measure of hypnosis in adults in the operating room and ICU.9 More recently it has been validated in the PICU.10 The BIS is an exciting new approach to EEG processing. It measures a state of the brain, representing the degree of alertness. It does not measure the concentration of a particular drug.11 A number of 100 on the BIS score indicates that the patient is fully awake, while a number less than 40 is suggestive of a deep hypnotic effect. A BIS value of less than 60 in surgical patients was not associated with a recall of intraoperative events.12 The use of the BIS monitor in adult surgical patients and in older pediatric patients has shown a reduction in anesthesia requirements and a shorter recovery time. The BIS monitor has been studied in several adult ICU populations. These studies have shown a correlation between the BIS score and a variety of sedation scores.13
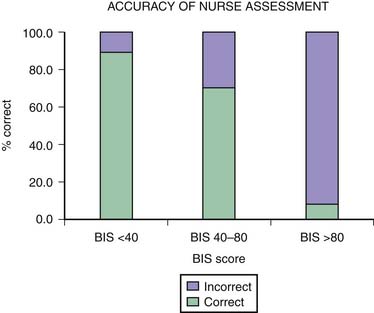
One of the main difficulties with clinical sedation scoring systems is their inability to assess depth of sedation in the patient receiving neuromuscular blocking agents (NMBAs). Patients who require NMBAs in the operating room are considered to be at increased risk of awareness during anesthesia.14 This problem also exists for the sedated patient with paralysis in the PICU. It is well known that the clinical signs of inadequate anesthesia or sedation are not reliable,15 and many other reasons account for alterations in the heart rate, blood pressure, perfusion, and pupillary responses in the PICU patient. In a study using the BIS in pediatric patients with paralysis,16 researchers found that in more than 8% of the sedation assessments in which patients were thought to be adequately sedated by the bedside nurse, their BIS scores were greater than 80 (Figure 123-1). This score reflected a significant risk of awareness. The BIS correlates well with the Ramsay scale in the sedated child and may be a useful monitor to prevent inadequate sedation in a child with paralysis.
Although the BIS monitor is used in many institutions, the question of whether its use can prevent awareness under anesthesia is still debated. A recent study enrolled 2000 adults whose anesthesia was either titrated to a BIS score of less than 60 or by the end-tidal inhalational concentration of the anesthetic agent to at least 0.7 minimum alveolar concentration (MAC).17 Postoperatively all patients were interviewed to assess their intraoperative awareness. This study found two cases of awareness in each group. The MAC values in both groups were the same. The BIS was greater than 60 in one case of awareness. Although the BIS monitor was not able to reduce this low incidence of awareness, the level of anesthesia between the groups was very similar. This study was severely underpowered to show any benefit. However, the combination of end-tidal monitoring and BIS monitoring may be helpful in reducing intraoperative awareness.
Other processed EEG sedation assessment monitors are now available. The SNAP IITM monitor18 uses a different spectrum of EEG frequency analysis. Little difference between the BIS and the SNAP IITM monitors has been shown thus far. Currently little experience has been reported with the SNAP IITM monitor in pediatric patients.
Opioids and Analgesia in the Pediatric Intensive Care Unit
Sedation in the PICU is most commonly achieved with a mixture of opioids and benzodiazepines (BZDs). Although many synthetic and naturally occurring opioids exist, morphine is considered the agent against which others are compared. The primary source of morphine is opium obtained from the opium poppy (Papaver somniferum), which also produces alkaloids such as codeine, thebaine, papaverine, and noscapine. Opiates are substances derived from opium; the term opioid also describes substances derived from opiates (e.g., oxycodone) but also includes substances that are created synthetically but have properties that are similar to those of opiates (e.g., fentanyl and methadone) and endogenous ligands. The terms often are used interchangeably because the pharmacologic effects fall into the same category. Opioids are agonists at various opioid receptors, for which several endogenous ligands exist. There are three major classes of receptors: mu (μ), kappa (κ), and delta (δ). The opioid receptors possess the same general structure of an extracellular N-terminal region, seven transmembrane domains, and an intracellular C-terminal tail structure. Subtypes of each receptor (e.g., μ1, μ2) exist (Table 123-3), as do less well-characterized opioid receptors ε, λ, τ, and ξ.
Table 123–3 Classification of Opiate Receptors and Subtypes
| Subtype | Prototypic Drugs | Actions |
|---|---|---|
| Mu1 | Opiates and most opiate peptides | Supraspinal analgesia including periaqueductal gray matter, nucleus raphe magnus, and locus coeruleus |
| Prolactin release | ||
| Acetylcholine turnover in brain | ||
| Catalepsy | ||
| Mu2 | Morphine | Respiratory depression |
| Dopamine turnover in brain | ||
| Gastrointestinal tract transit | ||
| Most cardiovascular effects | ||
| Delta | Enkephalins | Spinal analgesia |
| Dopamine turnover | ||
| Kappa | Dynorphin | Spinal analgesia |
| Inhibition of antidiuretic hormone | ||
| Sedation | ||
| Sigma | N-allynormetazocine | Psychotomimetic effects |
Modified from Baresh PG, Cullen BF, Stoelting RK et al, editors: Clinical anesthesia, ed 2, Philadelphia, 1992, JB Lippincott.
Most of the therapeutic and adverse effects can be accounted for by agonist activity at the μ-receptor, which is responsible for analgesia, respiratory depression, pupillary constriction, and euphoria. At the cellular level, μ-receptor activation alters ionic permeability to K+, causing hyperpolarization and depression of excitability in the neuronal system. Associated effects on cholinergic, adrenergic, serotonergic, and dopaminergic neurotransmitter systems are seen within the central nervous system (CNS). These receptors are found at multiple sites along pain pathways including the spinal cord, midbrain, thalamus, and the cortex. At the spinal cord level, pain reflexes (nociceptive) are depressed by receptors in the substantia gelatinosa, which are mostly presynaptic and inhibit the release of substance P from C-fiber nerve terminals and account for the effectiveness of intrathecally and epidurally administered opioids. In the midbrain the analgesic effect is mediated in the periaqueductal gray matter through ascending fibers and also descending fibers that modulate the function of the dorsal horn. Acetylcholine, γ-aminobutyric acid (GABA), norepinephrine, and serotonin also are involved in these pain-modulating pathways. Peripheral opioid receptors also have been shown and can be expressed in response to inflammation.19 The intraarticular injection of morphine produces analgesia following arthroscopy through activation of opioid receptors located on white blood cells.20
The endogenous ligands for the opioid receptors are the enkephalins, endorphins, and dynorphins. They have a morphine-like effect that can be specifically antagonized by the μ-receptor antagonist naloxone. The endomorphins have potent analgesic and gastrointestinal (GI) effects. At the cellular level, they activate G proteins ([35S] GTP gamma-S binding) and inhibit calcium currents.21 Pro-opiomelanocortin is the precursor for β-endorphin (as well as adrenocorticotropic hormone and melanocyte-stimulating hormone). β-Endorphin, itself very active, also includes the amino acid sequence for met-enkephalin, although the main precursor is proenkephalin A, which contains four copies of met-enkephalin and one copy of leu-enkephalin. The met-enkephalin sequence also gives opioid activity to a number of other larger peptides. Proenkephalin B (prodynorphin) gives rise to the dynorphin series and contains three leu-enkephalin sequences. Local application of these endogenous substances to the brain provides effects that are similar to those of opiates. They do not function as analgesics because the administration of naloxone does not cause pain in the normal state. They are released during periods of sustained pain, stress, or activity to modulate physiologic pathways, including those involved with pain. Therefore they are probably important to the physiologic condition of the patient in the ICU.
Specific Opioid Agonists
Morphine
Morphine is an opiate, and its primary therapeutic actions are sedation and analgesia; anxiolysis and euphoria also may occur. These four therapeutic effects may be exploited to the benefit of the patient. These actions are mediated through the periaqueductal gray matter, the ventromedial medulla, and the spinal cord. The reduction of nociceptive reflexes occurs all over the body, even below a completely transected spinal cord. In addition to increasing the sensory threshold for pain, morphine may decrease the hurting aspect (or unpleasantness) of pain. A patient given morphine may say something such as, “I have just as much pain, but it doesn’t distress me as much.” It blunts most types and intensities of pain, although some forms of neuropathic pain are relatively resistant. The resulting analgesia may be potent enough to abolish diagnostic symptoms and signs. The sedative effects reduce higher cortical function, cause difficulty in concentration, and cause a sense of drowsiness and dream-filled sleep. Higher doses will cause a state of unconsciousness or coma. The rate of respiration is reduced with a resultant fall in minute ventilation despite an accompanying increase in depth of breathing. This effect is associated with a decreased responsiveness to carbon dioxide (CO2) and is additive to the decreased CO2 response seen during sleep. In some circumstances respiratory drive may be restricted to hypoxic stimulation of the carotid chemoreceptors; this is the most serious dose-related adverse effect of morphine. It can occur at doses used clinically for analgesia. In general, all opiates produce the same degree of respiratory depression when given in equipotent doses and for any given level of analgesia. Opioids do not have anticonvulsant properties, whereas meperidine (and its metabolite normeperidine) may lower the seizure threshold.
Another CNS effect of morphine is pupillary constriction due to a central effect on the oculomotor nucleus. Nausea results from stimulation of the chemotrigger zone; however, opioids also depress the vomiting center, so the final effect is unpredictable. Nausea and vomiting are much more frequent in ambulatory patients than in patients confined to a hospital bed. Stress-related endocrine responses can be modified by morphine. It decreases the release of several hormones including adrenocorticotropic hormone, antidiuretic hormone, prolactin, growth hormone, and epinephrine. The neuroendocrine stress response that is normally seen with trauma and surgery may be blunted. Itching may be caused by histamine release, but it also may be due to opiate receptor activation in the spinal cord.22
Morphine’s effects on smooth muscle cause constipation. It reduces the intestinal propulsion activity through its central and peripheral effects. The central effects may be mediated by the vagus nerve. The direct smooth muscle relaxation and the increased local cholinergic transmission can be partly reversed by naloxone. This decreased motility is the basis of several over-the-counter antidiarrheal preparations including diphenoxylate, a μ-agonist that does not cross the blood-brain barrier and thus acts as a peripheral opioid agonist. Morphine also causes an increase in biliary tract tone, which may cause biliary colic, as well as increased tone in the bladder detrusor muscle and vesical sphincter. Urinary retention is common with opioids and occurs in 55% of children receiving spinally administered opioid and 20% receiving intravenous (IV) opioid.23
Morphine has been studied extensively in term and preterm neonates. Glucuronidation is present in term babies and in many preterm ones. The half-life of morphine, however, is 2 hours in children, 6.5 hours in term neonates, and 9 hours in the preterm child because of reduced clearance. Volume of distribution did not vary with age.24 Morphine causes histamine release and can cause peripheral vasodilatation. Infused at analgesic doses, it has little effect on the cardiovascular system, but skin flushing is not uncommon with rapid IV administration. The histamine-releasing potential should be considered in patients with asthma, especially during an acute exacerbation, and in patients with unstable cardiovascular systems for whom safer alternatives, such as fentanyl, exist.
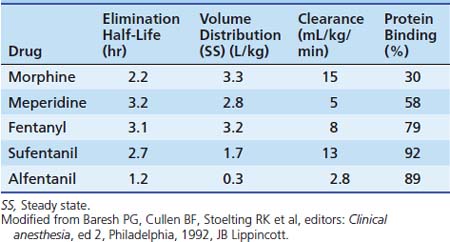
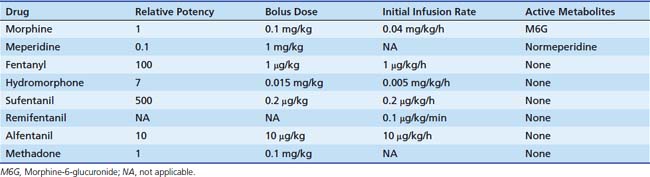
Dosing recommendations in the ICU include a bolus dose of 0.05 to 0.1 mg/kg and an infusion of 0 to 30 μg/kg/h. Fifty percent of these doses should be used if the patient is younger than 3 months of age. The pharmacokinetics of various opiates is outlined in Table 123-4. All opiates are weak bases and are moderately ionized at pH 7.4. Oral morphine is effective but undergoes hepatic first-pass metabolism, which is variable among patients. The oral dose for acute pain is two to five times the IV dose, while with long-term use the oral dose is 1.5 to 2.5 times the IV dose.
Morphine is metabolized to morphine-3-glucuronide (M3G) and M6G in the liver. M3G is the major metabolite and has little morphine-like activity, although some research has suggested that M3G may be associated with an antinociceptive effect, accounting for failure of analgesia during long-term use.25 In contrast, M6G is many times more potent than morphine itself.
Tolerance, defined as an increase in the dose required to create the same response, is a potential problem with all opiates. Tolerance is mainly limited to the depressant actions of morphine, including analgesia, respiratory depression, anxiolysis, and drowsiness. Tolerance of morphine’s inhibition of bowel motility and pupillary constriction is minimal. The mechanism of tolerance appears to involve the degree and duration of both μ- and κ-receptor occupancy. It appears more rapidly after continuous infusion, and cross-tolerance to other opiates is common, although anecdotal evidence suggests that when opioids are switched, a dose reduction may be possible because cross-tolerance sometimes appears incomplete.26 Receptor downregulation also may occur, as well as altered metabolism with an increased M3G/M6G ratio. Simultaneous blockade of N-methyl-D-aspartate receptors has been shown to be effective in reducing the development of tolerance.27 Clinical tolerance appears uncommon with an exposure of less than 3 days, but after prolonged administration, doses 10 to 20 times that which would cause respiratory arrest in nontolerant patients may be tolerated.
Meperidine
Meperidine has one tenth the potency of morphine. Compared with other common opioids, meperidine has more CNS excitatory effects including tremors, muscle spasm, myoclonus, psychiatric changes, and seizures. These effects may be due to a central serotoninergic effect.28 It is metabolized by the liver to normeperidine, which is twice as toxic as meperidine and has a longer half-life (15 hours). Normeperidine accumulation is enhanced in patients with an induced cytochrome P450 system. Meperidine has a shorter duration of action (2 to 3 hours) and has a more rapid onset because of its increased lipid solubility compared with morphine. Meperidine is unique among opioids because of its local anesthetic properties, which are capable of providing surgical spinal analgesia.29 A small dose (0.125 to 0.25 mg/kg) of meperidine may be used to treat postoperative shivering.
Fentanyl
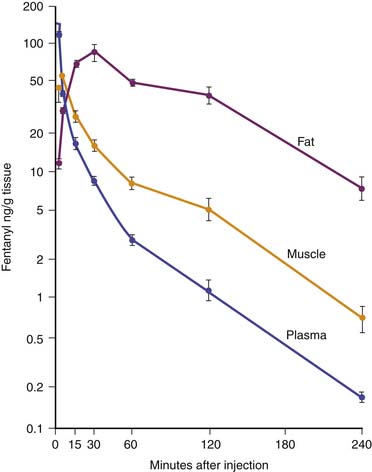
Fentanyl is one of the most commonly used opiates in the ICU. It is a synthetic derivative of meperidine without many of its unwanted side effects. It is a potent μ-agonist and is 100 times more potent than morphine. It has a rapid onset and cessation because of its high lipid solubility (Figure 123-2). Fentanyl may be administered by several routes, including IV, intramuscular (IM), transmucosal,30 and subcutaneous (SC) when venous access is inadequate. Skeletal muscle rigidity (which can occur with all synthetic opiates) is well described in the literature. It is mediated through the CNS and is an idiopathic response usually associated with a large bolus dose (≥5 μg/kg). It improves with the administration of NMBAs and is reversible with naloxone. Fentanyl has limited cardiovascular effects. Moderate bradycardia is the most common hemodynamic effect. Fentanyl does not cause histamine release. Dosing in the ICU is either by bolus (1 to 2 μg/kg) or infusion (1 to 4 μg/kg/h with higher doses as tolerance develops). The short duration of effect of a single dose of fentanyl is not due to metabolism but rather to rapid redistribution. Maximum brain concentration after a bolus is achieved within 90 seconds. Then, because of rapid redistribution, the plasma level falls by 50% in 30 minutes, and the result is a clinical duration of effect of a single dose of approximately 30 minutes. Fentanyl then accumulates in fat, where it is stored and slowly released with a longer elimination half-life of about 4 hours (longer than morphine). Marked respiratory depression occurs within 120 seconds, and a single dose of 5 μg/kg will cause apnea in 50% of patients. Also, fentanyl is metabolized by the liver to nor-fentanyl and hydroxy fentanyl derivatives, both of which are thought to be inactive. In the operating room, high-dose fentanyl is commonly used for cardiac anesthesia and for anesthetization of other unstable patients. A loading dose of 50 μg/kg, followed by 0.5 μg/kg/min, will occupy all opioid receptors and produce a state of anesthesia. Many cases of awareness with patients under anesthesia have been documented, however, even when these high doses of fentanyl were used.
Codeine
Codeine has a chemical structure and effects that are similar to those of morphine and is commonly used as an oral medication for cough suppression or mild to moderate pain relief. A large part of its effects are due to the metabolism of codeine to morphine. Ten to twenty percent of patients lack a metabolic pathway to convert codeine to morphine, which results in an unpredictable effect. Dosing is 0.5 to 1 mg/kg. Constipation is a major adverse effect, and some patients report having a vague peculiar or unpleasant feeling when they take codeine. This drug can be habit-forming. It can be given orally, IM, or rectally. Rapid IV use may result in cardiovascular collapse. Rectally administered codeine has been shown to have as rapid an onset as IM codeine, but it yields lower peak levels in children.31 Codeine has been the analgesic of choice by neurosurgeons because of the belief that pupillary signs are maintained with use of this drug. Morphine has been shown to be a more effective analgesic, however, in patients with head injuries.32
Remifentanil
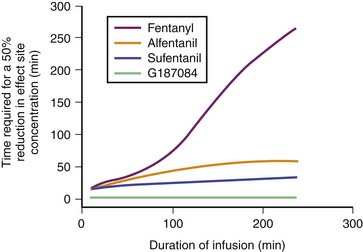
Remifentanil is one of the newest synthetic opiates available. It was designed to be metabolized by plasma esterases to provide a short half-life. It is a potent μ-agonist with mild κ and δ effects. It is substantially more potent than fentanyl. It is supplied as a white lyophilized powder that contains glycine (it should not be used for epidural or spinal analgesia). The metabolism is by nonspecific esterases not affected by pseudocholinesterase deficiency. The metabolite, a weak μ-agonist, is excreted by the kidney. The kinetics of remifentanil is different from those of most opiates used in the ICU. It has a short half-life that is due to metabolism rather than to redistribution. Therefore remifentanil has what is known as a context-sensitive half-life. The elimination half-life for remifentanil is about 8 minutes. With an infusion of remifentanil, the half-life does not increase but remains constant. With opiates such as fentanyl and alfentanil (Figure 123-3), the clinical effect half-life increases with time until it reflects the elimination half-life of between 2 and 4 hours.
Kinetics reported for neonates are similar to those reported for adults. The continuous infusion rate depends on the degree of sedation/analgesia required (0.1 to 0.5 μg/kg/min for sedation; 0.75 to 2 μg/kg/min for balanced anesthesia; 4 μg/kg/min for loss of consciousness). Remifentanil has effects on the cardiovascular system that are similar to those of fentanyl. Remifentanil causes a mild bradycardia and a slight decrease in blood pressure,33 which may be prevented with glycopyrrolate. No histamine release occurs. Remifentanil is a potent respiratory depressant. For spontaneous respiration, a low continuous infusion dose (without a bolus) should be used (0.1 μg/kg/min). Sedation can be effectively managed by continuous infusion without the need for a bolus because of the short half-life. An increase or decrease of infusion rate is rapidly reflected by a change in the degree of sedation, which is important to note. Most other opiate sedatives require bolus dosing to achieve a rapid change in effect. This type of dosing is neither appropriate nor needed for remifentanil.
Remifentanil has the usual opiate adverse effects; however, because of the short half-life, they have only brief clinical effect. Remifentanil may prove to be a safe and effective choice for PICU sedation in patients with severe renal or hepatic disease; however, the potential exists for glycine accumulation in patients with renal failure. It is an option only for those who require overnight ventilation or for those patients in whom a rapid awakening may be required for neurologic assessment. Remifentanil has been shown to reduce cerebral oxygen use and reduce cerebral blood flow if the CO2 is maintained in a normal range.34 Remifentanil is currently an expensive option and should not be considered for every patient. Also, because of its short duration, the postoperative patient may need an alternative analgesic after extubation. Rapid development of opiate tolerance with remifentanil has been described in healthy volunteers35 and also when used in the ICU setting. This rapid tolerance has also been described in postoperative scoliosis patients36; however, the increased morphine requirements described probably reflect the initial postoperative need to achieve an adequate morphine blood level rather than any acute tolerance.
Tramadol
Tramadol is an opiate analgesic that relieves pain by binding to opiate receptors and by inhibiting the reuptake in the CNS and spinal cord of norepinephrine and serotonin, two pain-modifying neurotransmitters. Tramadol does not have antiinflammatory effects. Its use is indicated in cases of moderate to severe pain.37 Despite being a narcotic-like agent, the Food and Drug Administration (FDA) has not classified tramadol as a controlled substance. Dosage (not FDA approved for patients younger than 16 years) is an initial oral dose of 1 to 2 mg/kg every 6 hours and should not exceed 6 mg/kg/day. An IV preparation is available outside of the United States. Patients with a creatinine clearance less than 30 mL/min should not receive a dose more often than once every 12 hours, with a maximum dose of 3 mg/kg/day. The dose for patients with cirrhosis or hepatic dysfunction is 1 mg/kg every 12 hours. Patients undergoing dialysis can receive their dose on the day of dialysis because only 7% of the drug is removed by the process. The adverse effects of tramadol most often involve the CNS and the GI tract. Patients may become dependent on tramadol. Abuse is possible, and it should not be given to opiate-dependent patients. Seizures have been seen in patients receiving high single oral doses of 10 mg/kg; this danger is even greater in patients with epilepsy and in anyone taking monoamine oxidase inhibitors and neuroleptic agents that lower the seizure threshold. Respiratory depression may occur if the recommended dosage is consistently exceeded or if another centrally acting depressant drug (e.g., alcohol) or an anesthetic is given concurrently. Because of the possibility of withdrawal symptoms, patients should not abruptly discontinue use of tramadol. Tramadol is not a useful drug for sedative action.
Table 123-5 provides conversion doses for some commonly used oral opiates. A summary of IV doses of different opiates is provided in Table 123-6.
Table 123–5 Dose Equivalents: Short Acting
| Drug | Oral | Parenteral |
|---|---|---|
| Morphine | 0.5 mg/kg q4h | 0.15 mg/kg q3h |
| Hydromorphone | 0.1 mg/kg q4h | 0.02 mg/kg q4h |
| Codeine | 4 mg/kg q3h | |
| Hydrocodone | 0.5 mg/kg q3h | |
| Oxycodone | 0.5 mg/kg q3h | |
| Meperidine | 5 μg/kg q2h | 1.5 mg/kg q2h |
| Fentanyl | 1.5 μg/kg q2h |
Opiate Antagonists
Several opiate antagonists are available. The most commonly used is naloxone, which is a specific and sensitive receptor antagonist of all opiate receptors. Dosing can be either low dose (1 μg/kg) or high dose in an emergency situation (10 μg/kg). If the drug cannot be administered intravenously, then it can be given intramuscularly, intranasally, or into the midventral surface of the tongue. When naloxone is being used for a long-acting agonist, an infusion may be necessary because its half-life is only 30 to 81 minutes (mean of 64 ± 12 minutes). In neonates, the half-life has been reported as 3.1 ± 0.5 hours; however, this prolonged effect is likely to be offset by a concomitant increase in the duration of action of the opioid for which the naloxone is given. No effect is seen in the healthy patient in the absence of administered opioids; however, in the setting of sepsis in the ICU, a vasopressor effect may occur, presumably because of an interaction with endogenous opioids released in response to stress. Nalmefene, a longer-acting antagonist, can be given through IV, IM, and SC routes. It has a redistribution half-life of 41 minutes and a terminal half-life of 10.8 hours in adults and somewhat less in children. Thus reappearance of the antagonized opioid is unlikely if it is given in an adequate dose.38
Incidental Pain Syndromes in the Pediatric Intensive Care Unit
In addition to the techniques used to sedate children to facilitate their PICU management, many children will have pain related to their underlying condition. Many different options are available for controlling pain in the pediatric patient (Box 123-1). The pharmacologic management of pain should follow the traditional World Health Organization analgesic ladder, which begins with a nonopioid analgesic such as a nonsteroidal antiinflammatory drug or acetaminophen, followed by a weak opioid such as hydrocodone added to the nonopioid, and then moves on to a strong opioid such as morphine or hydromorphone as needed. When taken orally, a sustained-release preparation is often useful once the dose requirement has been determined. The dose requirement of a strong opioid is variable in the patient taking opioids for a prolonged period, and failure to appreciate this variability is a common cause for therapeutic failure. In addition, once a dose requirement is known, the analgesic should be given to preempt pain rather than to relieve pain as required. At each level of analgesic use, the addition of adjuvant medications should be considered. Adjuvant drugs fall into six groups: antidepressant, anticonvulsant, neuroleptic, steroid, stimulant, and local anesthetic. Of the tricyclic antidepressants, nortriptyline is available in a liquid form. The tricyclic antidepressants are indicated for neuropathic pain, particularly when the patient describes a burning pain. Also useful for neuropathic pain are the anticonvulsant agents gabapentin, pregabalin, and carbamazepine. These drugs often work best when the pain is described as shooting or lancinating. Neuropathic pain may result from tumor invasion, vincristine therapy, cytomegalovirus infection, or human immunodeficiency infection. Neuroleptic drugs, including chlorpromazine and trimeprazine, may be useful in the management of nausea, anxiety, and pruritus. Steroids benefit mood, inflammation, nausea, appetite, nerve swelling/entrapment, and vasculitis. When opioid sedation is interfering with quality of life, a stimulant such as an amphetamine may restore energy and alertness while allowing ongoing analgesia from the opioid. Sometimes pain can be managed by local anesthetic, placed by peripheral nerve block, topically, or as a neuraxial block.
Patient controlled analgesia (PCA) has become the mainstay of postoperative pain relief in children because of its efficacy and safety. However, its use is limited by the child’s ability to understand how to use the PCA pump. Proxy PCA (PCA-P) has been used for younger children or those with cognitive impairment.39 The use of the PCA by the nurse or the caregiver may override the “safety net” that the PCA has. The use of PCA-P has been associated with greater need for rescue interventions; however, it is often used in sicker children. When PCA-P is used, careful evaluation and rigorous monitoring is needed.
Sickle Cell Crisis
Sickle cell disease differs from cancer and acquired immune deficiency syndrome in that intermittent episodes of severe pain occur, requiring urgent intensive treatment. A good review of this subject has been published.40 Chronic pain also may be present because of long-term tissue and bone damage from periods of ischemia during past crises, including persisting myocardial ischemia. Patients may be receiving long-acting opioids or may have had repeated exposure to opioids with past crises. Patients with a sickle cell disease crisis that involves the chest or brain are likely to be admitted to the PICU. Chest crises result from the sickling of erythrocytes in the pulmonary vasculature and result in hypoxia to the rest of the body and local lung damage. The systemic hypoxia worsens the crisis and is thus self-perpetuating. Chest radiograph changes may be late, and an associated paralytic ileus may be present. Poor pulmonary function may discourage the practitioner from using adequate opioid analgesics out of concern for worsening the hypoxia. However, it is important not to underestimate the need for pain relief and to appreciate that past opioid exposure may have resulted in tolerance to opioids. If IV access is difficult to obtain, morphine may be given subcutaneously or orally. Anecdotal success also has been reported with nebulized morphine.41
Opiate Tolerance
The use of opiate infusions in the ICU is associated with the potential for the development of tolerance or dependence.42 Iatrogenic withdrawal symptoms can occur if the opiates are discontinued abruptly. These effects have been shown to be related to the total dose and duration of fentanyl infusion. A fentanyl infusion of 5 days or a total cumulative dose of 1.6 mg/kg during the hospital stay was associated with a 50% chance of the development of narcotic withdrawal, whereas a fentanyl infusion of 9 days or longer or a total cumulative fentanyl dose of 2.5 mg/kg or more during the hospital stay had a 100% incidence of withdrawal.43 The rising plasma fentanyl levels caused by increased dosing suggested that increased metabolism or clearance was not responsible for the development of tolerance. A study of patients in a PICU to determine the degree of opiate tolerance has shown a significant increase in opiate dosing required for adequate sedation.44 The opiate infusion increased by about 80% per week for the first 3 weeks of opiate use. No difference in the rate of opiate increase was found with respect to age of the patient, postoperative status, mode of ventilation, and paralysis.
For patients considered to be at risk of withdrawal, several options are available. If circumstances allow, it is better to start the treatment for withdrawal prevention before the patient has symptoms and signs of withdrawal. Opiate withdrawal is not usually a serious medical problem; it is rarely life-threatening and is self-limited. However, treatment should be started early if possible for patient comfort. In a few circumstances the associated hypersympathetic state may not be good for the patient. The signs and symptoms of withdrawal are nonspecific, and other causes, such as infection, hypoglycemia, hypocalcemia, hyperthyroidism, and hypoxia, should be excluded. Because of the nonspecific nature of the symptoms of opiate withdrawal, several scoring systems have been described to aid with the diagnostic process. The Finnegan score is based on 31 variables and is lengthy. The Lipsitz score is shorter and easier to use than the Finnegan score. Both of these scoring systems, however, were devised for use with neonates, and several of the measurements are not appropriate for patients in the PICU. Currently no validated scoring system exists for assessing opiate withdrawal in the pediatric patient. In the limited number of articles in which opiate withdrawal in the PICU is evaluated, the authors have modified these scores (Table 123-7) in an attempt to provide an objective assessment of the patient.
Table 123–7 Signs and Symptoms of Opiate Withdrawal
| Sign/Symptom | Examples |
|---|---|
| Neurologic excitability | Sleep disturbances |
| Agitation | |
| Tremors | |
| Seizures | |
| Choreoathetoid movements | |
| GI disturbances | Vomiting |
| Diarrhea | |
| Autonomic dysfunction | |
| Hypertension (>150 mm Hg) | Tachycardia (>150 mm Hg) |
| Tachypnea (>40 beats/min) | |
| Fever (>38.58° C) | |
| Frequent yawning | |
| Sweating | |
| Goose flesh | |
| Mottling |
GI, Gastrointestinal.
Recently the assessment of a new score based on a similar set of signs and symptoms, the Withdrawal Assessment Tool,45 has been proposed. The bedside nurse reviews the patient’s chart for the previous 12 hours and performs a short assessment of the child’s level of agitation, as well as other signs/symptoms. This review is then followed by an assessment of the response to stimulus and also how quickly the child settles down after the stimulus. A score of 1 is assigned to each assessment (maximum, 12). A score of greater than 3 was associated with a greater likelihood of drug withdrawal. Although this score is new and has limited verification, it probably provides a better evaluation than just the clinician’s “bedside” opinion, especially in complex cases in which both opiate and BDZ withdrawal may occur together.
Several therapeutic options are available for the prevention and treatment of opiate withdrawal. Drugs from the same class are preferable. The FDA has approved methadone for opiate withdrawal. Other agents that may be useful include morphine, clonidine, dexmedetomidine, phenobarbital, paregoric, chlorpromazine, transdermal clonidine patch,46 and SC fentanyl. Paregoric contains morphine plus papaverine, noscapine, camphor (a CNS stimulant), ethanol (45%), benzoic acid (which competes with bilirubin-binding sites), and glycerin (which causes diarrhea). Paregoric has been used for neonatal withdrawal, but because of its composition, it may cause adverse effects. Chlorpromazine may be useful for GI adverse effects, but hypothermia and hypotension may occur. Haloperidol also may be of use, having minimal respiratory depression and no active metabolites. It also offers cardiovascular stability. Phenobarbital has been used for hyperactive behavior; however, it can cause significant CNS depression, it induces drug metabolism, and it is tolerance/dependence forming.
The convenience of the oral route, the less-frequent dosing because of its longer half-life, and the ease of calculating doses because of its equal potency to morphine make methadone attractive for use in the management of opiate withdrawal in children. However, a huge variability exists in recommendations regarding the methadone dose that should be used to prevent opiate withdrawal in children. Several factors are important in the dosing of methadone for conversion from fentanyl. After prolonged IV administration, fentanyl has a potency 100 times that of methadone; it has a metabolic half-life approximately one quarter that of methadone; and if given intravenously, it has a bioavailability 20% greater than orally administered methadone. In a study in which the effectiveness of a fentanyl-methadone conversion protocol was assessed, researchers found that giving 2.4 times the daily fentanyl dose as methadone prevented withdrawal symptoms.42 The methadone was given intravenously for 24 hours, and the fentanyl dose decreased by 50% on day 1 and by another 50% on day 2, and then it was discontinued. On day 3, the methadone was converted to oral dosing. The methadone was given intravenously initially. Because of its long half-life, oral dosing could take up to 5 days to reach a steady state. The duration of methadone requirement varied from 1 to 4 weeks, depending on the duration of opiate infusion. Methadone was being weaned by 3% to 15% per day with no signs of withdrawal. To date, there have been no published cases of respiratory arrest when methadone has been used for opiate weaning. However, it would appear prudent to initiate the conversion from fentanyl to methadone in the ICU environment in the event that problems arise and to ensure that an adequate dose is given. Once stabilized, the patient may be transferred to the floor and ultimately home, with a clearly described plan for decreasing the methadone dose over time. The weaning plan also should involve the home pediatrician so that patients have access to someone who is familiar with the process. In a follow-up of patients who had received methadone while in the ICU, 38% of patients were discharged home during the weaning process. No problems were associated with the weaning of methadone at home. Stigma regarding methadone use was not expressed by any of the parents.
The use of a clonidine patch also has been evaluated in the PICU. Clonidine has been shown to be effective in the management of nicotine, opiate, and alcohol withdrawal. It decreases sympathetic outflow from the CNS and has a synergistic effect for analgesia, both at the central and spinal level. In one report, eight patients were described after tracheal reconstructive operations. They required postoperative sedation and ventilation for 7 days, which put them at high risk for withdrawal.20 A clonidine patch was applied 12 hours before extubation, and the patients were weaned off the opiate. The dose used was approximately 6 μg/kg/day of clonidine, and the patch was left on for 7 days. One patch had to be removed because of hypotension. The patch seemed to be effective in preventing withdrawal. Use of the clonidine patch is attractive because of its noninvasive approach, which is desired. However, the use of a transdermal patch prevents titration of effect, and problems with bradycardia, hypotension, hypothermia, sedation, and dysrhythmia may occur.
A confounding issue in many publications and in the clinical management of opiate withdrawal is the potential for simultaneous BZD withdrawal. Most researchers have not been able to separate these two issues. The symptoms of BZD withdrawal differ from those of opiate withdrawal because the BZD symptoms generally include less sympathetic activation. BZD withdrawal symptoms are characterized by agitation and a movement disorder. If BZD withdrawal is a concern, low-dose lorazepam or diazepam may be added to the withdrawal management strategy (Table 123-8). A prospective study of BZD withdrawal47 following lorazepam infusion (up to 0.3 mg/kg/h) documented BZD withdrawal syndrome in approximately 25% of the children. This withdrawal occurred even when using a 6-day tapering of the lorazepam dose. All the children had been previously weaned off fentanyl infusions. No predisposing risk factors were found for BZD withdrawal with respect to BZD or opiate dosing or duration.
Rapid Opiate Detoxification
Reports have been made of rapid opiate detoxification in the ICU. These procedures have used a form of deep sedation (often with use of propofol or another anesthetic agent) to facilitate opioid withdrawal in patients addicted to the recreational use of opiates.48 The patients are given high doses of opiate antagonists to displace all opiates from the receptors and then heavy sedation is initiated to reduce the occurrence and effects of the sympathetic stimulation observed with short-term opiate withdrawal. These procedures have been safely performed in the ICU; however, there have been several reports of complications49 when these procedures were not performed with full ICU support. Currently the effectiveness and safety of 1-day opiate detoxification is still an area of debate.50 If used, however, it should be combined with an established long-term support plan to optimize long-term success.
In the PICU, deep sedation with propofol has been used to facilitate rapid opiate weaning of ventilator-dependent patients.51 The use of propofol for up to 3 days allowed a reduction of fentanyl dosing from 24 to 9 μg/kg/h (a 65% reduction). No signs or symptoms of opiate withdrawal were noted, and metabolic acidosis did not develop. Opiate antagonists were not used for this rapid weaning process. However, concern has been raised regarding the long-term administration of propofol, especially in the PICU patient, given the development of the propofol infusion syndrome.
Benzodiazepines
BZDs are among the most commonly used agents for sedation in the ICU. They augment the function of the GABA type A (GABAA) receptor at the postsynaptic membrane. This pentameric protein controls a chloride channel, the opening of which leads to an inhibitory effect due to hyperpolarization of the cell membrane.52,53 Benzodiazepines bind to BZD receptors, which in the CNS are usually found as part of the GABAA receptor, enhancing the effect of endogenous GABA.54 Peripheral BZD receptors55 are not usually associated with the GABAA receptor but are a binding site for diazepam and midazolam. These 18-kDa proteins are associated with regulation of cellular proliferation, immunomodulation, porphyrin transport, heme biosynthesis, and anion transport. In particular, they seem important in the regulation of steroid synthesis and apoptosis, and they have a significant effect on the hypothalamic-pituitary-adrenal axis.56 These latter effects may be pertinent to the physiologic care of patients in the ICU.
BZD receptors are bound by a family of endogenous peptides called endozepines, which have similar effects to the BZDs.57,58 The expression of this diazepam-binding inhibitor may be relevant to the development of dependence not only on BZDs but also on alcohol and opioids59 and may therefore be relevant in the drug dependence commonly seen in patients in the PICU who are given these agents continuously. Naturally occurring BZDs have been detected with structures similar to those used clinically.60 Subsets of GABAA receptors have been shown to have different effects. Type 1 receptors were responsible for sedation and anterograde amnesia, whereas type 2 receptors mediated anxiolysis. It may be possible to develop selective subtype receptor agonists to provide anxiolysis without sedation, amnesia, or dependence.
The general pharmacologic effects of BZDs are sedation, anxiolysis, euphoria (limbic system), reduced skeletal muscle tone (through spinal BZD receptors), anticonvulsant properties, and neuroendocrine effects. They impair acquisition and encoding of new information, providing anterograde amnesia. They do not have any analgesic properties. They have little direct effect on ICP. Their effects are dose dependent. Patient cofactors including age, concurrent disease, and any cosedation therapy influence responses to BZDs. Paradoxical reactions are reported in which agitation rather than calming is observed.61 In healthy patients, BZDs have few cardiovascular adverse effects, but in a sick, intensive care population, profound cardiovascular depression may be observed occasionally. BZDs should be used judiciously until the patient response is known.62 Midazolam has been most often associated with this effect,63 and research in dogs has shown both negative inotropy and chronotropy, especially when the sympathetic response has been abolished.64 Clinical use is largely encompassed by discussion of the pharmacologic properties of diazepam, midazolam, and lorazepam.
Specific Benzodiazepines
Diazepam
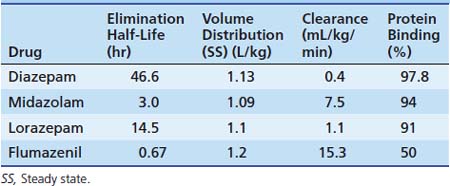
The first widely used BZD in the ICU was diazepam. Because of its low solubility in water, it is available in the IV or IM form dissolved in propylene glycol. This formulation causes a significant amount of pain and thrombophlebitis with peripheral IV use. A lipid emulsion that has fewer adverse effects is available in the United Kingdom. Diazepam is inexpensive and is effective for short-term sedation; in such cases, accumulation is less of a concern. Diazepam may be given orally because it has good absorption, but absorption tends to be erratic when it is given rectally or intramuscularly. It is highly lipid soluble with a long half-life (24 hours). Metabolism by oxidative biotransformation generates several hypnotically active metabolites with an elimination half-life that may be longer than diazepam, including oxazepam (half-life, 10 hours) and n-dimethyldiazepam (half-life, 93 hours). Delayed recovery has been reported in neonates after they received diazepam, possibly because of the long half-life of dimethyldiazepam.65 Prolongation of effects occurs in patients when clearance is reduced because of hepatic dysfunction and when metabolism is inhibited by drugs such as cimetidine and omeprazole.
Midazolam
Midazolam is an imidazobenzodiazepine. It has a short elimination half-life of 2 hours and is water soluble, which means that IV injection is nonirritating. Because of these factors, it has become popular in ICUs for sedation by infusion. Intranasally (0.2 mg/kg), midazolam has proven to be as effective at controlling febrile seizures as IV diazepam (0.3 mg/kg).66 It has extensive first-pass metabolism and provides less reliable results when given PO, although this route is often successfully used for premedication of children before general anesthesia in doses of 0.5 to 0.75 mg/kg (maximum, 20 mg). It is available in a pleasant-tasting cherry syrup and is effective in 10 to 15 minutes, providing up to 1 hour of adequate anxiolysis, although residual hangover effects may persist.67 Rectal and sublingual administration has been described.
Midazolam is about eight times more potent than diazepam, with starting dose recommendations of a bolus dose of 0.05 to 0.1 mg/kg68 and an infusion of 1 to 6 μg/kg/min. Midazolam is metabolized by the cytochrome P450 system subfamily IIIA (nifedipine oxidase), polypeptide 4 (CYP3A4),69 to hydroxymidazolam (63% potency) and hydroxymidazolam glucuronide (9% potency). Because of the high degree of protein binding (94% protein bound), the free level can be significantly changed with interactions because of the protein binding, which also may occur with heparin. Hepatic or renal failure increases the free fraction by two to three times, and its effect also can be prolonged by the accumulation of active metabolites.70 The half-life of midazolam in patients in the ICU may be prolonged compared with that in healthy patients.71 With short-term infusions (<12 hours), it retains a rapid recovery; however, with increased duration of use, the recovery becomes prolonged. Its clearance may be reduced by several commonly used ICU drugs, including calcium channel blockers, erythromycin, and triazole antifungal agents.72
Lorazepam
Lorazepam is an alternative water-soluble agent that is well absorbed after both oral and IM administration.73 It produces sedation for 4 to 8 hours after a single dose. Lorazepam has a slower onset than does midazolam. The elimination half-life is about 14 hours. Metabolism is by glucuronyl transferase, not the cytochrome P450, and there are no active metabolites. This metabolism is unaffected by cimetidine or phenobarbital, which only affects oxidative metabolic pathways. Sodium valproate may inhibit its metabolism.74 In persons with advanced liver disease, these phase II glucuronidation reactions are better preserved, and the increased half-life seen is due to increases in the volume of distribution rather than to reduced clearance. In patients with renal failure, prolonged half-life is also due to reduced protein binding because clearance is unchanged. No change in metabolism occurs with aging or critical illness. In a comparison of infusions of midazolam and lorazepam, the recovery characteristics were found to be significantly different. In patients receiving lorazepam, it took an average of 260 minutes to return to baseline, whereas in patients receiving midazolam, it took more than six times longer to return to baseline. Lorazepam may be administered by bolus (0.05 to 0.1 mg/kg every 2 to 4 hours) or by infusion (0.05 mg/kg/h). Lorazepam is slightly less expensive than is midazolam.75 It has been recommended as the BZD of choice for long-term sedation because of its more predictable recovery profile in sick patients in the ICU. Lorazepam for IV use has propylene glycol as a carrier. Risk of a metabolic lactic acidosis exists because of the metabolism of this carrier. Cases of fatal metabolic acidosis from propylene glycol have been reported in neonates taking a particular vitamin preparation. Several other potential ICU drugs may use propylene glycol as a carrier, including some IV preparations of phenytoin and phenobarbital, nitroglycerin, digoxin, and etomidate. Reports of propylene glycol toxicity in adults who received multiple propylene glycol infusions have been made.76 Care should be taken when lorazepam is infused in patients who receive these other medications. In patients in the PICU, propylene glycol levels have been shown to correlate with the dose of lorazepam received; however, no metabolic abnormalities were detected.77 Hemodialysis has been used successfully in the management of the lorazepam-associated propylene glycol toxicity.78
The metabolisms of different BZDs are intertwined with each other. Most of the agents require an oxidative process first with potentially active compounds before glucuronidation and excretion. The pharmacokinetics for different BZDs is shown in Table 123-8.
Tolerance and Dependence to the Benzodiazepines
Tolerance for and dependence on BZDs can occur as with opiates in the PICU.79 This effect is not all due to receptor number downregulation.80 Withdrawal symptoms may be avoided with a slow taper of the medication of 10% per day or by substituting a long-acting oral agent such as diazepam. Acute withdrawal symptoms may include anxiety, insomnia, nightmares, seizures, psychosis, and hyperpyrexia. A postmidazolam infusion phenomenon has been described that includes poor social interaction, decreased eye contact, and a decreased interest in the surroundings. The patient may exhibit choreoathetotic movements with dystonic posturing that can persist for 2 to 4 weeks but will resolve with no sequelae.
Flumazenil
Flumazenil is an imidazobenzodiazepine and is a specific competitive antagonist of the BZD receptor. It has no effect on other drugs such as barbiturates, ethanol, or other GABA-mimetic agents. Flumazenil reverses the hypnotic and sedative effects of BZDs. It has a half-life of approximately 1 hour after a single IV bolus. In patients with hepatic impairment, its half-life and clearance are prolonged, and a significant increase (>50%) of free drug occurs because of reduced plasma protein binding. Renal failure has little effect on the pharmacokinetics of flumazenil. It is indicated for the complete or partial reversal of the central sedative effects of BZDs. Contraindications include patients who have a known hypersensitivity to BZDs, patients with epilepsy who are receiving treatment with BZDs, and persons who have overdosed with a tricyclic antidepressant. The use of flumazenil is often associated with mild to moderate tachycardia and hypertension.
In cases of multiple drug overdose, the use of flumazenil remains controversial. It often is overused in the emergency setting without due concern for potential adverse reactions81 because of the potential toxic effects (e.g., cardiac arrhythmias or convulsions) of other psychotropic drugs ingested. The toxicity of tricyclic antidepressants becomes apparent as the effects of BZDs are antagonized. Patients should be evaluated for the signs and symptoms of a tricyclic antidepressant overdose; an electrocardiogram (ECG) may be helpful in determining the risks involved.
The dosing information for pediatric patients is limited. The initial suggested dose is 0.01 mg/kg (maximum, 0.2 mg) with incremental doses of 0.005 to 0.01 mg/kg (maximum, 0.2 mg) given every minute up to a maximum cumulative dose of 1 mg. The lower doses are suggested for sedation reversal and the higher doses for BZD overdose. Infusions at 0.05 to 0.01 mg/kg/h have been used.82 The use of flumazenil in sedated patients in the ICU should be tempered by the potential for an unrecognized BZD dependence, which would increase the risks of adverse effects. If its use is required, then a carefully titrated dose would be appropriate. The half-life of flumazenil is much shorter than that of some of the BZDs it may be counteracting (see Table 123-8). The use of an infusion may be necessary because resedation has been reported after single-bolus use.83 However, this requirement should not preclude the use of flumazenil in an ICU setting.84 Flumazenil has been used for the reversal of moderate sedation. In the pediatric population, although it was well tolerated, it was not shown to significantly reduce recovery time.85 Because flumazenil has a limited duration with the potential for resedation after discharge from medical care, an appropriate period of observation is required before discharge. A study in which researchers monitored the effects of flumazenil after sedation indicated that some of the residual effects of midazolam were still present after reversal.86 Flumazenil also has been used to treat a paradoxical midazolam reaction87 and has been shown to be effective in the management of hepatic encephalopathy or hyperammoneamia.88 A Cochrane Collaboration review of articles pertaining to flumazenil use demonstrated a short-term improvement in hepatic encephalopathy. However, no improvement in recovery or survival was documented. No serious adverse effects were noted.
Chloral Hydrate
Chloral hydrate is a widely used oral hypnotic/sedative agent. It has been used for sedation for radiographic procedures, for EEGs, and in many different health care locations. It was first synthesized in 1832 and used in 1869 as a hypnotic agent. Shortly after, reports of acute and chronic toxicity were published.89 In 1910, it was labeled as the most dangerous of hypnotics even though heroin and opium were in common use at that time. The addition of ethanol potentiates its effect (street name “Mickey Finn”). It has been used to control agitation in the intensive care nursery and to treat sleep difficulties in older patients.
Chloral hydrate is rapidly and completely absorbed from the GI tract and is immediately converted into the active component, trichloroethanol (TCE), by alcohol dehydrogenase.90 The plasma levels peaks at 30 to 60 minutes. TCE is 45% protein bound. TCE undergoes glucuronidation with some oxidation to trichloroacetate (TCA). The half-life of TCE is 8 to 12 hours, while that of TCA is 67 hours. In infants and neonates this may be increased by a magnitude of three to four. With multiple dosing, a significant potential exists for accumulation. TCA can displace bound bilirubin from albumin. Its actions include CNS depression with drowsiness and sleep in less than an hour. With an overdose, the patient falls into a deep stupor or coma, and the pupils change from contracted to dilated. At therapeutic levels, the blood pressure and respiratory rate are unaffected. Chloral hydrate has little hangover effect. It has several effects on the cardiovascular system including decreased myocardial contractility, a shortened refractory period, and an increased sensitivity of the heart to catecholamines. It also has effects on mucous membranes. Irritation can cause gastritis, nausea, and vomiting. With overdose, a severe hemorrhagic gastritis with gastric necrosis and esophagitis has been described. Chloral hydrate and ethanol interfere with one another’s metabolism through competition for alcohol dehydrogenase. Also, ethanol inhibits the conjugation of TCE, and TCE inhibits the oxidation of ethanol. Coumadin activity may be increased by chloral hydrate. Chloral hydrate is synergistic with other sedative agents. In children receiving amphetamine-based medication, chloral hydrate is contraindicated because there have been rare reports of arrhythmias. The reversal of chloral hydrate with flumazenil has been described; however, a report of ventricular tachycardia with this combination also has been made.
Signs of toxicity are usually noted within 3 hours of dosing. Paradoxical excitement also has been described in 6% of patients. There is some evidence that chloral hydrate may be genotoxic and carcinogenic. Mice studies have shown that a single-dose exposure can result in an increased risk of hepatic carcinomas and adenomas.91 Chloral hydrate overdose produces a clinical picture that is similar to acute barbiturate poisoning. Ataxia, lethargy, and coma occur within 1 to 2 hours. Also, a pearlike odor may be noted. Cardiovascular instability poses the main threat to life. Severe arrhythmias including atrial fibrillation, supraventricular tachyarrhythmia, ventricular tachyarrhythmia, torsades de pointes, and ventricular fibrillation have been described. Chronic use can cause a dependence syndrome. Also, chloral hydrate is not detectable in the blood. TCE levels are measurable, but they are not useful for clinical management, although they can be helpful for retrospective diagnosis. The management of toxicity includes evaluation and monitoring at a medical facility if an amount greater than 50 mg/kg or an unknown amount has been ingested. Two capsules may cause significant toxicity in a toddler, so there is little room for error in the history. Charcoal with intubation should be considered if significant toxicity is suspected. Standard antiarrhythmic management is often unsuccessful, although esmolol, overdrive pacing, and hemoperfusion have been tried.








