Chapter 70 Renal Pharmacology
Kidney Function and Drug Disposition
The pharmacologic effect of a therapeutic agent is determined by the amount, and time, that the active drug component (e.g., parent drug or drug metabolite) is present at the site of action and the responsiveness of the target organ to the drug. Only the free, unbound form of a drug can exert a pharmacologic effect through interaction with receptors. The disposition of a drug after administration is determined by the formulation and route of administration, the rate and extent of absorption from the site of administration, the extent of distribution in the body fluids and tissues, the rate and extent of metabolism, and the rate and route of elimination. Drug disposition and response are further influenced by the genetic, physiologic, and pathologic constitution of the ill child.1,2 Pharmacokinetics is the mathematic expression of drug disposition, and pharmacodynamics describes the magnitude of response to a drug. The determination of pharmacokinetic parameters is an invaluable tool for designing individualized drug-dosing regimens in children with kidney failure or critical illnesses that may alter drug disposition (see also Chapter 116).
Although decreased kidney elimination of drugs and drug metabolites is the most obvious consequence of altered kidney function, kidney failure or the associated coexisting conditions may affect drug absorption, distribution, and metabolism (Table 70-1).3–5 The mechanisms that govern drug removal by the kidneys are glomerular filtration, tubular secretion, and tubular reabsorption. These processes are altered in children with kidney disease, as well as other organ dysfunction. Each of the kidney’s one million nephrons consists of a tuft of capillaries (glomerulus) enveloped by an epithelial-lined capsule (Bowman’s space) that drains into a contiguous tubular system. As blood flows through the glomerular capillaries, fluid and small solutes, including drugs and drug metabolites not bound to plasma proteins, pass through the glomeruli into Bowman’s space. The volume of water and accompanying solute that is filtered through the glomeruli per unit time is the glomerular filtration rate (GFR) and is the most important measure of kidney function (see Chapter 69). The GFR is estimated with the measurement of the rate that the kidney removes a substance from the blood (e.g., renal clearance). The measured substance may be an endogenous compound (e.g., creatinine [Cr] or cystatin C), an exogenous compound that is specifically administered to measure the GFR (e.g., inulin, isotope, or iohexol), or a compound primarily eliminated by glomerular filtration that is administered as part of clinical care (e.g., gentamicin).6 The clearance of Cr corrected for body surface area is the most common method used to estimate the GFR. Creatinine clearance (CCr), expressed in mL/min/1.73 m2, is calculated with the measurement of the amount of Cr in an accurately timed urine collection and a mid collection plasma Cr.
Table 70–1 Potential Alterations of Drug Distribution in Kidney Failure
| PK PARAMETER | EFFECT | PROPOSED MECHANISM |
|---|---|---|
| Absorption | ↓ | |
| Distribution | ↑ | |
| Metabolism | ||
| Excretion | ↓ |
CYP 450, Cytochrome P450; GFR, glomerular filtration rate; GI, gastrointestinal; N/V, nausea and vomiting; PK, pharmacokinetic.
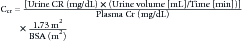
CCr is low at birth and rapidly increases during the first 2 weeks of life, followed by a steady rise until adult values are reached by 8 to 12 months.7,8
Although not as accurate as a timed urine collection, for children aged 1 to 18 years, CCr can be quickly estimated with the measurement of the child’s serum Cr and length with the following equation9:
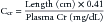
where the plasma creatinine is calibrated to the isotope-dilution mass specrometry method of measuring serum creatinine. It is important to understand that the normal relationships among serum Cr, length, and GFR are altered in disturbances of Cr biosynthesis (e.g., muscular disease and malnutrition) or in clinical settings where the serum Cr is rapidly changing (e.g., acute renal failure, recovery from renal failure, and dialysis). In these situations, a timed urine collection is required for an accurate estimate of the GRF.
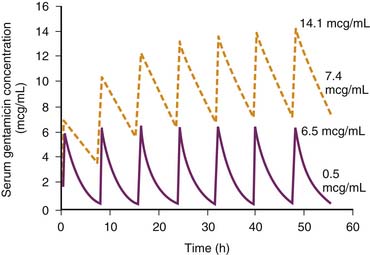
The total amount of drug eliminated from the body by the kidneys is a composition of the amount of drug filtered across the glomeruli, the amount actively secreted into the filtrate by the renal tubules, and the amount reabsorbed by the renal tubules. Because albumin and other large molecules do not pass through the glomeruli, large drugs and drugs bound to plasma proteins normally do not pass through the glomeruli and are not effectively removed by glomerular filtration but may be efficiently eliminated by the kidney through renal tubular secretion (e.g., furosemide). For drugs and drug metabolites that are primarily eliminated by glomerular filtration, the rate of elimination mirrors kidney function (CCr). As such, when kidney function declines, the reduced drug elimination results in drug accumulation in the body. For example, gentamicin is an aminoglycoside that is eliminated primarily by glomerular filtration with an elimination half-life of around 2 hours in children and adults with normal kidney function and 4 to 12 hours in infants because of the well-characterized developmental immaturity of kidney function.7,8 A 75% reduction of the GFR (e.g., CCr = 30 mL/min/1.73 m2) in a 5-year-old child receiving intravenous (IV) gentamicin will prolong the elimination half-life to 8 hours. Unless adjustments are made in the dosing regimen to account for the decreased kidney elimination, gentamicin will accumulate to toxic serum concentrations (Figure 70-1).
The active renal tubular secretion of drugs and drug metabolites by relatively nonspecific anionic and cationic transport systems in the proximal tubule can contribute substantially to the amount of drug eliminated by the kidney. The renal tubular secretion of a drug may be inhibited by other drugs or endogenous substrates that use the same nonspecific transport systems. For example, probenecid blocks the tubular secretion of many drugs by the organic anion transporter and is of clinical utility as an adjuvant to antibiotic therapy and the prevention of drug-induced nephrotoxicity.10–12 For example, the coadministration of probenecid and penicillin-type antibiotics results in higher and more prolonged serum penicillin levels because of the inhibition of penicillin secretion by the renal tubule. The increased penicillin exposure may enhance the therapeutic efficacy of penicillin.13,14 Nephrotoxicity is the major dose-limiting adverse effect of the antiviral agent cidofovir and is in part mediated by the proximal tubular transport of cidofovir by the organic anion system. Coadministration of probenecid restricts the proximal renal tubular uptake of cidofovir and decreases the incidence of nephrotoxicity.11 Reabsorption is the passive diffusion of non-ionized (noncharged) drug from the filtrate into the renal tubular cell. Basic urine (e.g., urine pH >7.5) favors the ionized form of acidic drugs and limits reabsorption, whereas reabsorption of basic drugs is enhanced in basic urine because the non-ionized form of the drug is favored. Urinary alkalinization is used to enhance the renal elimination of salicylates and possibly barbiturates in overdose situations (see Chapters 105 and 106).15
Drug Dosing in Kidney Disease
Drugs and drug metabolites that are predominately eliminated by the kidney will accumulate to higher serum drug concentrations in patients with decreased kidney function if adjustments are not made to the drug-dosing regimen. A systematic approach to individualized drug therapy in children with kidney failure will ensure maximal therapeutic efficacy and minimize toxicity (Box 70-1). The first step in designing a rational individualized dosing regimen is an estimation of the child’s kidney function with measurement of the CCr as described in the previous section. The next step is to evaluate the effect of kidney failure on the drug disposition characteristics for all of the drugs prescribed to the child. Reference books such as the Pediatric Dosage Handbook,16 Physicians Desk Reference, and Micro Medex are excellent beginning sources for information about drug disposition in kidney failure. Unfortunately, because there is little pharmacokinetic information about drug disposition in children with kidney failure, estimates from adult patients with kidney failure are cautiously used, keeping in mind the potential changes in drug disposition that occur with development.17 The most important information for designing an optimal dosing regimen is the amount of drug that is eliminated by the kidneys in persons with normal kidney function. If one assumes that drug protein binding, distribution, and metabolism are not greatly altered in kidney failure, an assumption that is likely true for most drugs,5 then a dosage adjustment factor (Q) can be estimated:
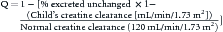
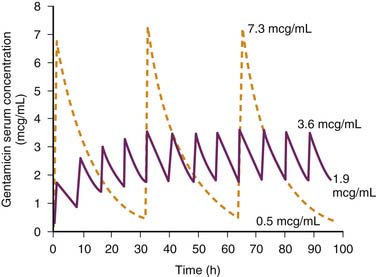
Once the need for a dosage adjustment has been established and the adjustment factor (Q) calculated, the best method of adjustment, whether it be a change in the size of the dose or the length of the dosing interval, is selected on the basis of the known relationships between the peak and trough drug concentrations and clinical response or toxicity. Figure 70-2 shows the concentration-time profiles for two different IV gentamicin dosing regimens in a child with a measured CCr of 30 mL/min/1.73 m2. About 95% of gentamicin is excreted unchanged in the urine, and the dosage adjustment factor is calculated as:
< div class='tao-gold-member'>
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


