CHAPTER 9 Regulation of Hemostasis and Thrombosis
Hemostasis
Necessary Components
Extravascular Tissues
When an injury disrupts a blood vessel, it allows blood to contact extravascular cells and matrix. Extracellular matrix proteins, such as collagen, fibronectin, thrombospondin, and laminin, interact with adhesive receptors on blood platelets and support formation of the initial platelet plug at the site of injury. Perivascular tissues also express significant levels of tissue factor (TF).1,2 Exposure of TF to blood initiates the process of thrombin generation on the surfaces of adherent platelets and ultimately leads to stabilization of the initial platelet plug in a fibrin clot (i.e., secondary hemostasis). Different tissues express different complements of matrix components and procoagulants. The tissue environment plays a role in determining the intensity of the procoagulant response to an injury.
Platelets
Membrane receptors for collagen and other subendothelial and extravascular matrix proteins are present on the platelet membrane and mediate binding of unactivated platelets at sites of injury.3–5 Platelet binding is also mediated by von Willebrand factor (vWF) bridging between collagen and the platelet receptor glycoprotein (GP) Ib. These receptor binding events also transmit an activation signal to the platelets. Full platelet activation also requires stimulation by thrombin, however, which is produced as the coagulation reactions are initiated. The platelet surface receptor for fibrinogen, GPIIb/IIIa, rapidly changes conformation from an inactive to an active form on platelet activation.6 This change in conformation allows platelet aggregates to be stabilized by binding to fibrinogen even before conversion to fibrin begins. Platelet activation also initiates the synthesis of prostaglandins and thromboxanes—compounds that modulate platelet activation and promote vasoconstriction.7
Coagulation Proteins
The vitamin K–dependent factors include factors II (prothrombin), VII, IX, and X. These factors each have a structural domain in which several glutamic acid residues are post-translationally modified to γ-carboxyglutamic acid (Gla) residues by a vitamin K–dependent carboxylase.8 The vitamin K cofactor is oxidized from a quinone to an epoxide in the process. A vitamin K epoxide reductase cycles the vitamin K back to the quinone form to allow carboxylation of additional glutamic acid residues. The negatively charged Gla residues bind calcium ions. These binding interactions hold the Gla-containing proteins in their active conformation. The calcium-bound form of the Gla-domain is responsible for mediating binding of the coagulation factors to phospholipid membranes. Lipids with negatively charged head groups, particularly phosphatidylserine, are required for binding and activity of the Gla-containing factors.
The carboxylation process is inhibited by the anticoagulant warfarin, which competes with vitamin K for binding to the reductase.9 Inhibition by warfarin results in the production of undercarboxylated forms of the vitamin K–dependent proteins, which are nonfunctional.
Factor IIa (thrombin) is a little different from the activated forms of the other vitamin K–dependent factors. Its Gla domain is released from the protease domain during activation. It no longer binds directly to phospholipid membranes. It also does not require a cofactor to cleave fibrinogen and initiate fibrin assembly, or to activate platelet receptors. Factor IIa that escapes the vicinity of a hemostatic plug can bind, however, to a cofactor on endothelial cell surfaces, thrombomodulin.10 After binding to thrombomodulin, factor IIa can no longer activate platelets or cleave fibrinogen. Instead, it triggers an antithrombotic pathway by activating protein C on the endothelial surface.
Proteins C and S are also vitamin K–dependent factors. They do not act as procoagulants, but rather as antithrombotics on endothelial surfaces.11 Protein C is the zymogen of a protease, whereas protein S has no enzymatic activity, but serves as a cofactor for activated protein C. The activated protein C/protein S complex cleaves and inactivates factor Va and factor VIIIa, preventing propagation of thrombin generation on normal healthy endothelium.
TF is also a cofactor, but is structurally unrelated to any of the other coagulation factors. Instead, it is related to one class of cytokine receptors.12 This lineage emphasizes the close evolutionary and physiologic links between the coagulation system and the other components of the host response to injury. Rather than circulating in the plasma as do the other coagulation factors, TF is a transmembrane protein.13 TF serves as the cellular receptor and cofactor for factor VIIa. It is primarily expressed on cells outside the vascular space under normal conditions, although monocytes and endothelial cells can express TF in response to inflammatory cytokines. The factor VIIa/TF complex can activate factor IX and factor X, and is the major initiator of hemostatic coagulation.13
Another group of related proteins are the contact factors—factors XI and XII, prekallikrein, and high-molecular-weight kininogen. These proteins share the feature of binding to charged surfaces. The only one of this group that is needed for normal hemostasis is factor XI.14 The other contact factors may play a role, however, in thrombosis in some settings. Factor XI is a zymogen that can be activated to a protease by factor XIIa, but is likely activated primarily by thrombin during the hemostatic process. Factor XIa activates factor IX.
Fibrinogen provides the key structural component of the hemostatic clot. Two small peptides, fibrinopeptides A and B, are cleaved from fibrinogen by thrombin, and the resulting fibrin monomer polymerizes into a network of fibers. The fibrin polymer is stabilized further when it is cross-linked by activated factor XIII. Factor XIIIa is a transglutaminase that is activated by thrombin coincident with fibrin formation.15
Thrombin plays a key role in activating procoagulant and anticoagulant factors and triggering formation of fibrin. In addition, thrombin has cytokine-like activities that bridge the transition between hemostasis, inflammatory/immune responses, and wound healing. Thrombin is truly a multifunctional molecule that affects the host response to injury at many levels.
Even before the structure and function of the various factors had been defined, their interactions had been studied during plasma clotting. In the 1960s, two groups proposed a “waterfall” or “cascade” model of the interactions of the coagulation factors leading to thrombin generation. These schemes were composed of a sequential series of steps in which activation of one clotting factor led to the activation of another, finally leading to a burst of thrombin generation.16,17 At that time, each clotting factor was thought to exist as a proenzyme that was activated by proteolysis. The existence of cofactors without enzymatic activity was not recognized until later. The original models were subsequently modified as information about the coagulation factors accumulated and eventually evolved into the Y-shaped scheme shown in Figure 9-1. The “cascade” model shows distinct intrinsic and extrinsic pathways that are initiated by factor XIIa and the factor VIIa/TF complex. The pathways converge on a “common” pathway at the level of the factor Xa/factor Va (prothrombinase) complex.
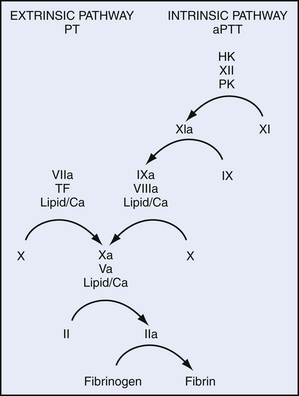
The numbering of the coagulation factors does not follow their order in the cascade. The coagulation factors were numbered roughly in the order in which they were discovered. Because many workers had described the same molecules under different names, designating them with roman numerals seemed the fairest way to reconcile the nomenclature confusion.18
Process of Hemostasis
Having all the right ingredients is not enough to ensure an effective hemostatic process. Cellular interactions are crucial to directing and controlling hemostasis. Normal hemostasis is impossible in the absence of platelets. In addition, TF is an integral membrane protein, and its activity is normally associated with cells, but platelets have little TF activity. Interactions between at least these two types of cells are necessary. Because different cells express different levels of procoagulants and anticoagulants and have different complements of receptors, it is logical that simply representing the cells involved in coagulation as phospholipid vesicles overlooks the active role of cells in directing hemostasis. Hemostasis in vivo can be conceptualized as occurring in a stepwise process, regulated by cellular components,19 as described subsequently.
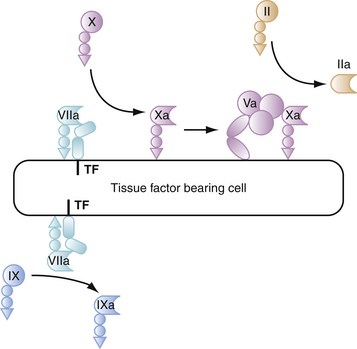
Step 1: Initiation of Coagulation on Tissue Factor–Bearing Cells
The process of thrombin generation is initiated when TF-bearing cells are exposed to blood at a site of injury. TF is a transmembrane protein that acts as a receptor and cofactor for factor VII. When bound to TF, zymogen factor VII is rapidly converted to factor VIIa through mechanisms not yet completely understood, but that may involve factor Xa or noncoagulation proteases. The resulting factor VIIa/TF complex catalyzes activation of factor X and activation of factor IX. The factors Xa and IXa formed on TF-bearing cells have very distinct and separate functions in initiating blood coagulation.20 The factor Xa formed on TF-bearing cells interacts with its cofactor, factor Va, to form prothrombinase complexes and generate small amounts of thrombin on the TF cells (Fig. 9-2). The small amounts of factor Va required for prothrombinase assembly on TF-bearing cells are activated by factor Xa,21 activated by noncoagulation proteases produced by the cells,22 or released from platelets that adhere nearby. The activity of the factor Xa formed by the factor VIIa/TF complex is largely restricted to the TF-bearing cell because factor Xa that dissociates from the cell surface is rapidly inhibited by tissue factor pathway inhibitor (TFPI) or AT in the fluid phase.
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Vascular Complications after Percutaneous Coronary Intervention
Vascular Complications after Percutaneous Coronary Intervention
 Diagnosis and Treatment of Complications of Coronary and Valvular Interventions
Diagnosis and Treatment of Complications of Coronary and Valvular Interventions
 Pharmacologic Interactions in the CICU
Pharmacologic Interactions in the CICU
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


