Chapter 20 Regional Circulation
General Features
General Anatomy
The large arteries are elastic. Their media contain concentric lamellae of perforated elastic tubes cross-linked by transverse collagen and smooth muscle. The wall becomes stiffer when its smooth muscle contracts. Smaller arteries have fewer lamellae. The media are bounded by the internal and external elastic laminae. Arterioles are less elastic, have no lamellae, and have thin media with circular or spiral smooth muscle and inner and outer elastic laminae. Capillaries have thin walls and are nonmuscular, which is ideal for transport of materials into and from the tissues. Veins have medial muscle but have thinner walls relative to lumen diameter than do arteries. The vascular endothelium has important metabolic characteristics that may differ between vessel types (i.e., arteries vs. veins) and in different regions.1,2
Basic Physiology
In a model to explain the relation of arterial pressure to flow, the circulation is represented by two capacitance vessels separated by a resistance. A standpipe full of blood is allowed to discharge its contents into the arterial vasculature (the proximal capacitance). Blood flows across the resistance site (the arterioles), traverses the venous vasculature (the distal capacitance), and drains to a reservoir at some outflow pressure (Po), taken for this example to be atmospheric (Figure 20-1).
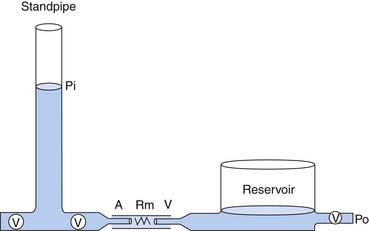
The pressure head of the system (Pi) is generated by the weight of the column of blood in the standpipe and is proportional to its height (Pi = blood column height in cm H2O). As the standpipe discharges, the height decreases and Pi decreases. This, in turn, decreases the rate of flow (Q) through the vasculature. Q decreases almost linearly with Pi until the column is quite low. Ultimately, flow will cease while there is still pressure in the standpipe. The pressure at which flow ceases is the critical closing pressure of the circulation (Pcc).3 Lower Pi is insufficient to maintain vessel patency and to permit continued flow (Figure 20-2).
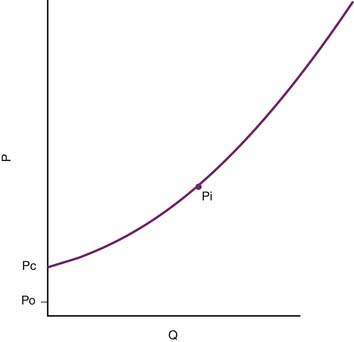
Figure 20–2 As the standpipe in Figure 20-1 discharges, Pi falls. Flow consequently slows and ultimately stops when Pi = Pc, the critical closing pressure of the circulation. Pi only reaches outflow pressure Po if Po ≥ Pc. P, Pressure; Q, rate of flow.

Venous Return and Cardiac Output
A second model illustrates the relationship of cardiac output to intrinsic mechanical properties of the systemic vasculature. In this model, the heart acts as a roller pump, creating a circulation much like that achieved during extracorporeal membrane oxygenation (Figure 20-3).
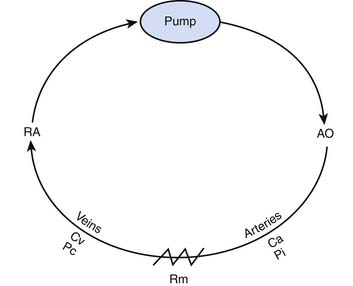
As Q is reduced, by turning down the roller pump, Pi falls and the volume of blood that resides in the arterial circuit also declines. This phenomenon allows more blood to reside in the veins, and venous pressure rises. As Q approaches zero, venous pressure approaches the mean circulatory pressure (Pm) (Figure 20-4). The importance of this model is that it can be used to illustrate the role of venous return as an independent determinant of cardiac output.
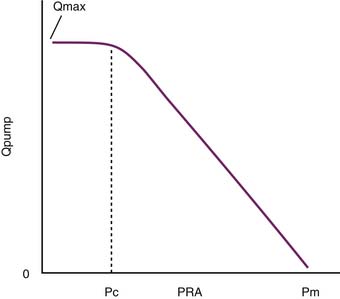
In its simplest form, cardiac output and central venous pressure of the intact circulation can be assumed to behave as illustrated in Figure 20-5. An increase in venous pressure distends the heart and elevates cardiac output. This behavior of the heart (see Chapter 19) can be superimposed on pressure flow characteristics of the vasculature. Venous return and cardiac output achieve equilibrium at one theoretical point for any given state of cardiac function (Starling curve) and simultaneous set of vascular characteristics (venous return curve). This imaginary point defines a unique equilibrium relation of cardiac output to venous pressure. This venous return curve is dramatically influenced by changes in blood volume, vascular capacitance, and vascular resistance.
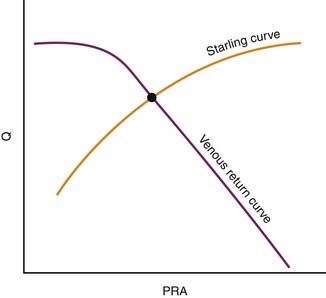
Transfusion elevates the maximal venous return that can be achieved without venous closure. Phlebotomy has the opposite effect. Because neither transfusion nor phlebotomy directly alters resistance or capacitance, the slope of the curve is not altered (Figure 20-6). Both interventions alter mean circulatory pressure because both interventions change blood volume.
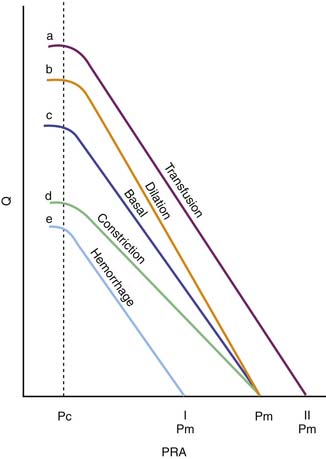
Isolated manipulation of incremental resistance would alter the maximum venous return attainable without venous collapse, yet it would not alter mean circulatory pressure. Changes in incremental resistance thus change the slope of the venous return curve (see Figure 20-6).
Critical Closing Pressure
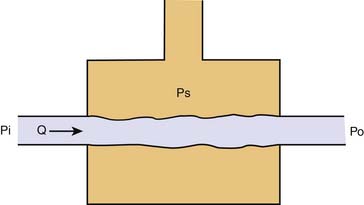
In many organs, as inflow pressure is lowered Q decreases, and it ceases at a pressure—the critical closing pressure (Pcc)—that is higher than venous pressure. The probable mechanism is the vascular waterfall or Starling resistor. In 1910 Jerusalem and Starling described a device that was designed to control afterload to the left ventricle and that made possible the study of cardiac contractility.4 The device consisted of a collapsible rubber tube traversing a pressurized glass chamber (Figure 20-7). When pressure surrounding the rubber tube exceeded the outflow pressure set by the reservoir, surrounding pressure opposed the flow of blood and became the true outflow pressure of the device. The physiologic counterpart of this phenomenon occurs in small vessels surrounded by tissue pressure. In the heart, for example, a Starling resistor effect occurs in extramyocardial coronary veins, although evidence also exists for critical closure of small arterioles. No one has yet demonstrated vessel closure directly, but this might not be necessary because as small vessels narrow when they are compressed, the wall becomes convoluted, and blood cells might become obstructed by the folds even when externally the vessel does not appear to be closed.
Autoregulation
In all organs, when inflow pressure is suddenly raised or lowered while oxygen consumption remains constant, flow rises or falls transiently but then returns to its former value; this phenomenon is termed autoregulation. Myogenic tonic response is in part responsible for this phenomenon, but it is not the only mechanism. Some investigators have shown that tissues have oxygen sensors that respond to transient increases or decreases in oxygen supply, and other studies indicate that the process is mediated by greater or lesser release of nitric oxide carried to the tissues by hemoglobin in the form of S-nitrosohemoglobin.5–7 Other locally produced gases such as hydrogen sulfide8 and carbon monoxide also may play a role.9,10 Evidence indicates that some autoregulatory mechanisms are specific to individual microcirculations (e.g., macula densa signaling in the renal circulation).
Distensibility and Compliance
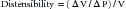
where V is volume and P is pressure. Veins are much thinner than arteries and are about eight times more distensible. Multiplying distensibility by volume yields ΔV/ΔP, which is the definition of compliance. Because venous volume is usually more than three times arterial volume, venous compliance is about 20-fold to 30-fold greater than arterial compliance. As a result, whenever fluids are infused, the bulk of the fluid volume is accommodated in the veins.
Vascular Resistance
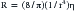
where R is resistance, l is tube length, r is the internal radius of the tube, and η is the fluid viscosity. Blood is not a Newtonian fluid, but this fact does not affect the accuracy of calculated vascular resistance much. However, vascular beds do contain many “tubes” in parallel. Thus for vascular systems a factor k is added that represents the number of vessels. The equation thus becomes:
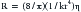
Local Regulatory Mechanisms
Vascular tone is strongly influenced by several mechanisms: (1) innervation and neural processes, (2) circulating endocrine and neuroendocrine mediators, (3) local metabolic products, (4) blood gas composition, (5) endothelial derived factors, and (6) myogenic processes.
Innervation and Neural Processes
Receptors responsive to neural products (e.g., norepinephrine and acetylcholine) are found throughout the circulation. Nevertheless, innervation and receptor distribution are organ specific, allowing rapid, patterned, coordinated redistribution of blood flow and an orchestrated response to events such as hypoxia, changes in posture, and hemorrhage. Although these receptors respond to circulating agonists (including adrenal epinephrine) as well as to those liberated locally, they generally are associated with innervation by autonomic nerves. In general, presynaptic α-adrenergic stimulation causes norepinephrine reuptake, whereas postsynaptic α-adrenergic stimulation causes norepinephrine release and vasoconstriction. β-Adrenergic stimulation generally causes vasodilation. Cholinergic stimulation (whether sympathetic or parasympathetic) generally causes vasodilation (see Chapters 25 and 117).
Local Metabolic Products
Local metabolic regulation of vasomotor tone provides an ideal homeostatic mechanism whereby metabolic demand can directly influence perfusion. The precise mechanisms underlying the coupling of blood flow with metabolic activity remain unclear. One theory holds that as the metabolic rate increases, so too does the formation of some vasodilating substance. Thus the regional vasculature relaxes, allowing more oxygen to be delivered in support of this work. As flow rises, the metabolites are washed out, restoring their concentration to normal. Adenosine, for instance, which accumulates locally when tissue metabolism is high and tissue oxygenation is marginal, causes pronounced vasodilation in the coronary, striated muscle, splanchnic, and cerebral circulations. Another example is potassium, which is released from muscle in response to increased work, ischemia, and hypoxia.11 Hypokalemia causes vasoconstriction, and hyperkalemia, within the physiological range, causes vasodilation.12,13
An increasing amount of data demonstrate the importance of the local redox state on the regulation of blood flow through the microcirculation. Reactive oxygen species such as superoxide, hydrogen peroxide, and peroxynitrite have been shown to influence normal regulatory processes and to participate in the pathophysiology of a wide array of cardiovascular disorders. For example, the rapid reaction of nitric oxide (NO) with the superoxide anion results in the formation of peroxynitrite, a potent oxidant. While peroxynitrite is known to have cytotoxic properties, under normal conditions peroxynitrite inhibits leukocyte adherence and platelet aggregation without evidence of cellular injury.14,15 In disease states, however, peroxynitrite can lead to protein nitration and deoxyribonucleic (DNA) damage. In addition, elevated levels of superoxide may decrease the bioavailability of NO, leading to abnormal vasomotion.16
Blood Gas Composition
Tissue levels of oxygen and carbon dioxide have been shown to reflect adequacy of perfusion and oxygen delivery.17 These blood gases are potent determinants of regional blood flow and have effects that differ from one region of the circulation to another. They also have a more general effect mediated by carotid chemoreceptors.
Endothelial-Derived Factors
The vascular endothelial cells are capable of producing a variety of vasoactive substances that participate in the regulation of normal vascular tone. These substances, such as NO, carbon monoxide (CO), hydrogen sulfide, and endothelin-1 (ET-1) are capable of producing vascular relaxation and/or constriction, modulating the propensity of the blood to clot, and inducing and/or inhibiting smooth muscle migration and replication (Figure 20-8).18,19 The understanding of the role of the vascular endothelium and the factors it produces in regulating blood flow in health and disease has resulted in several treatment strategies that target, mimic, or augment endothelial processes. Examples include inhaled NO for pulmonary hypertension, L-arginine supplementation for coronary artery disease and the pulmonary vasculopathy of sickle cell disease, phosphodiesterase inhibitors (i.e., sildenafil, which prevents the breakdown of cyclic guanosine 3′5′-monophosphate [cGMP]) for pulmonary hypertensive disorders, endothelin receptor antagonists for pulmonary hypertensive disorders and vasospasm following subarachnoid hemorrhage, and NO inhibitors for refractory hypotension resulting from sepsis.20–25 Indeed, many older therapies used to promote vascular relaxation, such as nitrovasodilators, affect endothelial function, a fact that until relatively recently was not appreciated.
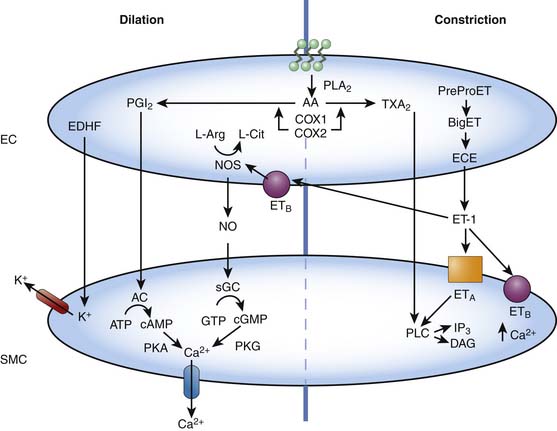
NO is a labile humoral factor produced by NO synthase from L-arginine in the vascular endothelial cell. NO diffuses into the smooth muscle cell and produces vascular relaxation by increasing concentrations of cGMP via the activation of soluble guanylate cyclase. NO is released in response to a variety of factors, including shear stress (flow) and the binding of certain endothelium-dependent vasodilators (such as acetylcholine, adenosine triphosphate [ATP], and bradykinin) to receptors on the endothelial cell. Basal NO release is an important mediator of both resting pulmonary and systemic vascular tone in the fetus, newborn, and adult, as well as a mediator of the fall in pulmonary vascular resistance normally occurring at the time of birth.26–29 Dynamic changes in NO release are fundamental to the regulation of all vascular beds.
CO is a labile humoral factor produced by the action of hemoxygenase on heme in many tissues, including endothelial cells. Hemoxygenase-1 is constitutive and hemoxygenase-2 is inducible. CO interacts with NO, is an independent stimulator of cGMP, relaxes smooth muscle, inhibits its replication, and has powerful antithrombotic and antiinflammatory effects. It is beginning to enter the field of clinical medicine.10,30
Hydrogen sulfide is produced in most tissues by a variety of mechanisms and may be the ultimate sensor that is stimulated by oxygen deficit or excess.8 Hydrogen peroxide may be even more important.
ET-1 is a 21-amino-acid polypeptide also produced by vascular endothelial cells.31 The vasoactive properties of ET-1 are complex, and studies have shown varying hemodynamic effects on different vascular beds. However, its most striking property is its sustained hypertensive action. In fact, ET-1 is the most potent vasoconstricting agent discovered, with a potency 10 times that of angiotensin II. The hemodynamic effects of ET-1 are mediated by at least two distinctive receptor populations, ETA and ETB. The ETA receptors are located on vascular smooth muscle cells and mediate vasoconstriction, whereas the ETB receptors may be located on endothelial cells and mediate both vasodilation and vasoconstriction. Individual endothelins occur in low levels in the plasma, generally below their vasoactive thresholds, which suggests that they are primarily effective at the local site of release. Even at these levels, they may potentiate the effects of other vasoconstrictors such as norepinephrine and serotonin.32 The role of endogenous ET-1 in the regulation of normal vascular tone is presently unclear.33 Nevertheless, alterations in ET-1 have been implicated in the pathophysiology of a number of disease states.34
Endothelial-derived hyperpolarizing factor (EDHF), a diffusible substance that causes vascular relaxation by hyperpolarizing the smooth muscle cell, is another important endothelial factor. EDHF has not yet been identified, although it is likely an epoxyeicosatrienoic acid, but current evidence suggests that the action of EDHF is dependent on K+ channels (Figure 20-8).35 Activation of potassium channels in the vascular smooth muscle results in cell membrane hyperpolarization, closure of voltage-dependent calcium channels, and ultimately vasodilation. Potassium channels are also present in endothelial cells. Activation within the endothelium results in changes in calcium flux and may be important in the release of NO, prostacyclin, and EDHF. Potassium channel subtypes include ATP-sensitive K+ channels, Ca2+-dependent K+ channels, voltage-dependent K+ channels, and inward-rectifier K+ channels.35
The breakdown of phospholipids within vascular endothelial cells results in the production of important byproducts of arachidonic acid, including prostacyclin (PGI2) and thromboxane. PGI2 activates adenylate cyclase, resulting in increased cyclic adenylate monophosphate production and subsequent vasodilation, whereas thromboxane results in vasoconstriction via phospholipase C signaling (Figure 20-8). Other prostaglandins and leukotrienes also have potent vasoactive properties.
Myogenic Processes
In 1902, Bayliss36 described an intrinsic increase in vascular tone in response to elevated intravascular pressure. This myogenic response results in alterations in vascular tone following changes in transmural pressure or stretch.37 This response is especially important at the arteriolar level and is thought to participate in regional autoregulation. Increases in intravascular pressure and/or stretch result in an increase in arteriolar smooth muscle tone, while decreasing pressures have the reverse effect. The precise mechanisms mediating this response are unclear, but a role for dynamic changes in intracellular Ca2+ and myosin light chain phosphorylation has been documented.38 More recent work has focused on the role of tyrosine phosphorylation pathways, ENaC, transient receptor potential channels, potassium channels, and alterations in Ca2+ sensitivity in this response.39–43 Moreover, the myogenic response varies between the regional circulations and between vessels within a given circulation.
Regional Circulations
Pulmonary Circulation
The morphologic development of the pulmonary circulation affects the physiologic changes that occur in the perinatal period. In the fetus and neonate, small pulmonary arteries have thicker muscular coats than similarly located arteries in the adult. This muscularity is, in large part, responsible for the pulmonary vascular reactivity and high resistance found in the fetus. In fetal lamb lungs fixed at normal in utero perfusion pressures, the medial smooth muscle coat is most prominent in the smallest arteries (fifth- and sixth-generation arteries; external diameter 20-50 μm). During the latter half of gestation, the medial smooth muscle thickness remains constant in relationship to the external diameter of the artery.44 Similar observations utilizing slightly different techniques have been made in human lungs.45,46 Within the first several weeks after birth, the medial smooth muscle involutes and the thickness of the media of the small pulmonary arteries decreases rapidly and progressively.47
Following this perinatal transition, the medial layers of the proximal pulmonary vascular bed are completely encircled by smooth muscle. Moving distally, muscularization becomes incomplete (arranged in a spiral or helix) and disappears completely from the most peripheral arterioles.46 In these arterioles, an incomplete pericyte layer is found within the endothelial basement membrane. Smooth muscle precursor cells reside in the nonmuscular portions of the partially muscular pulmonary arteries.48 Under certain conditions, such as hypoxia, these cells may rapidly differentiate into mature smooth muscle cells.48
Normal Fetal Circulation
In the fetus, normal gas exchange occurs in the placenta and pulmonary blood flow is low, supplying only nutritional requirements for lung growth and performing some metabolic functions. Pulmonary blood flow in near-term lambs is about 100 mL/100 g wet lung weight, representing between 8% and 10% of the total output of the heart.49 Pulmonary blood flow is low despite the dominance of the right ventricle, which in the fetus ejects about two thirds of the total cardiac output. Most of the right ventricular output is diverted away from the lungs through the widely patent ductus arteriosus to the descending thoracic aorta, from which a large proportion reaches the placenta through the umbilical circulation for oxygenation. In young fetuses (at about halfway through gestation), pulmonary blood flow is approximately 3% to 4% of the total combined left and right ventricular outputs of the heart (fetal cardiac output). This value increases to about 6% at about four fifths through gestation, corresponding temporally with the onset of the release of surface active material into lung fluid. This step is followed by another progressive slow rise in pulmonary blood flow, reaching about 8% to 10% near term.49 Fetal pulmonary arterial pressure increases with advancing gestation. At term, mean pulmonary arterial pressure is about 50 mm Hg, generally exceeding mean descending aortic pressure by 1 to 2 mm Hg.50 Pulmonary vascular resistance early in gestation is extremely high relative to that in the infant and adult, probably because of the low number of small arteries. Pulmonary vascular resistance falls progressively during the last half of gestation, new arteries develop, and cross-sectional area increases; however, baseline pulmonary vascular resistance is still much higher than after birth.50,51
Changes in the Pulmonary Circulation at Birth
After birth, with initiation of ventilation by the lungs and the subsequent increase in pulmonary and systemic arterial blood O2 tensions, pulmonary vascular resistance decreases and pulmonary blood flow increases by eightfold to tenfold to match systemic blood flow (300 to 400 mL/min/kg body weight). This large increase in pulmonary blood flow increases pulmonary venous return to the left atrium, increasing left atrial pressure. Then the valve of the foramen ovale closes, preventing any significant atrial right-to-left shunting of blood. In addition, the ductus arteriosus constricts and closes functionally within several hours after birth, effectively separating the pulmonary and systemic circulations. Mean pulmonary arterial pressure decreases, and by 24 hours of age, it is approximately 50% of mean systemic arterial pressure. Adult values are reached 2 to 6 weeks after birth (Figure 20-9).52,53
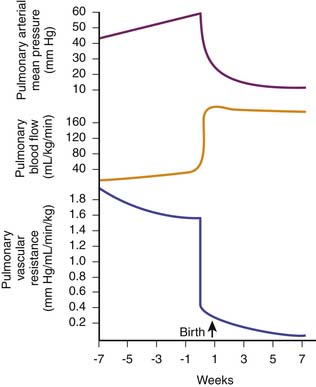
The decrease in pulmonary vascular resistance with ventilation and oxygenation at birth is regulated by a complex and incompletely understood interplay between metabolic and mechanical factors, which in turn are triggered by the ventilatory and circulatory changes that occur at birth. Physical expansion of the fetal lamb lung without changing O2 tension increases fetal pulmonary blood flow and decreases pulmonary vascular resistance, but not to newborn values.54 A very small proportion of this decrease relates to replacement of fluid in the alveoli with gas, which allows unkinking of the small pulmonary arteries, and to the changes in alveolar surface tension, which exert a negative dilating pressure on the small pulmonary arteries, maintaining their patency.55 Physical expansion of the lung also releases vasoactive substances such as PGI2, which increases pulmonary blood flow and decreases pulmonary vascular resistance in the fetal goat and lamb.56 There also is net production of PGI2 by the lung with the initiation of ventilation at birth.56 In addition, inhibitors of prostaglandin synthesis (such as indomethacin or meclofenamic acid) not only block PGI2 production but also attenuate the increase in pulmonary blood flow and decrease in pulmonary vascular resistance that occur with physical expansion of the fetal lung, although not the changes that occur with oxygenation.57 Therefore PGI2 (or perhaps, but less likely, another metabolite of arachidonic acid) plays an important role in the increase in pulmonary blood flow and decrease in pulmonary vascular resistance that occur in association with the mechanical component (stretch) of ventilation at birth.
Ventilation of the fetus without oxygenation produces partial pulmonary vasodilatation, whereas ventilation with air or oxygen produces complete pulmonary vasodilatation. The exact mechanisms of oxygen-induced pulmonary vasodilation during the transitional circulation remain unclear. The increase in alveolar or arterial O2 tension may decrease pulmonary vascular resistance either directly by dilating the small pulmonary arteries or indirectly by stimulating the production of vasodilator substances such as PGI2 or NO. In particular, NO has been implicated as an important mediator of the decrease in pulmonary vascular resistance at birth associated with increased oxygenation.58–63 However, the immediate decrease in pulmonary vascular resistance minutes after birth is not attenuated by NO inhibition. Therefore the decrease in pulmonary vascular resistance with initiation of ventilation and oxygenation consists of at least two components. First is partial pulmonary vasodilatation caused by physical expansion of the lung and the production of prostaglandins (PGI2 and PGD2). This process probably is independent of fetal oxygenation and results in a modest increase in pulmonary blood flow and a decrease in pulmonary vascular resistance. Second is a further maximal pulmonary vasodilatation associated with fetal oxygenation, which is not necessarily dependent on prostaglandin production. This process results in an increase in pulmonary blood flow and a decrease in pulmonary vascular resistance to newborn values. This latter pulmonary vasodilatation is likely caused by the synthesis of NO. Both components are necessary for the successful transition to extrauterine life. An additional mechanism by which vasodilatation occurs is related to the stimulation by increased shear forces of endothelial cells to produce both NO and PGI2. It is possible that after the initial fall in pulmonary vascular resistance because of another mechanism, this particular mechanism acts to maintain pulmonary vasodilatation.
Regulation of Postnatal Pulmonary Vascular Resistance
After the immediate postnatal state, the pulmonary circulation is maintained in a dilated, low-resistance state. Because the inflow pressure of the pulmonary circulation is quite low, a vertical gradation to the distribution of blood flow in the lung occurs. Hydrostatic pressure must be adjusted for vertical height above the left atrium, both at the inflow and at the outflow of every alveolar capillary unit. For example, given a pulmonary artery mean pressure of 20 cm H2O (zeroed at the level of the left atrium), an alveolar-capillary unit 12 cm above the left atrium will face an inflow pressure of only 8 cm H2O. A left atrial pressure of 5 cm H2O would generate no opposing outflow pressure to alveolar capillary units more than 5 cm above the left atrium. Critical closing pressure of postcapillary vessels therefore would set outflow pressure for a unit 10 cm above the left atrium. Were intrinsic vascular resistance identical throughout the lung, flow at any vertical height would be determined by hydrostatic driving pressure (inflow-outflow) and would be greatest at the base and least at the apex of the lung. West, Dollery, and Naimark64 reported that this phenomenon partitions the lung into three vertical regions (Figure 20-10). Zone I vessels are higher above the left atrium than pulmonary artery pressure (expressed in cm H2O) and are not perfused by the pulmonary artery. Zone II vessels lie above the height defined by the hydrostatic left atrial pressure but below the height of pulmonary artery pressure. These units are perfused in proportion to the driving pressure across them, which is approximately pulmonary artery pressure less vertical height (or critical closing pressure, whichever is higher). Zone III vessels lie at a vertical height less than outflow pressure expressed in cm H2O. Driving pressure across these units is independent of height because inflow and outflow pressures are comparably influenced by gravity. Zone I likely does not exist in a supine neonate.
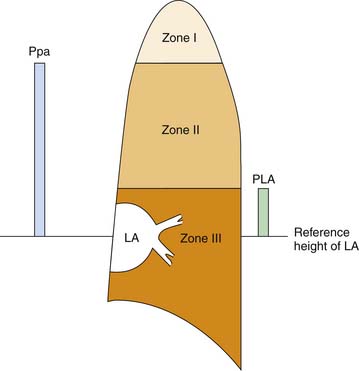
Because small pulmonary arteries course along with the branching airways and small pulmonary vessels are intimately related with alveoli, airway pressure can directly modulate pulmonary blood flow.65 Alveolar pressure can be loosely translated into surrounding pressure for alveolar vessels. Positive airway pressure applied to the lung can impinge on alveolar vessels whenever alveolar pressure exceeds the other determinants of outflow pressure. During positive pressure ventilation, outflow pressure of the pulmonary circulation may be determined predominantly by the mechanics of ventilation. The lung is partitioned into zones, but the distribution of flow becomes a complex function of alveolar pressure as well as left atrial and critical closing pressures.
To further complicate this view of the pulmonary circulation, lung volume and alveolar pressure both change during positive pressure ventilation. During inspiration, extraalveolar and corner vessels are dilated by radial traction, reducing their resistance to flow, whereas alveolar vessels narrow and elongate.66 During lung inflation, alveolar surface tension rises, diminishing the transmission of alveolar pressure to alveolar vessels.67 Evidence also indicates that, in the infant lamb, lung stretch may directly augment pulmonary vascular tone in a manner that is dependent on calcium flux and subject to calcium channel blockade using verapamil.68 In fact, it is clear that mechanical ventilation can have profound direct effects on the intact pulmonary circulation that depend on the waveform of airway pressure applied and not on mean airway pressure alone.69,70
In heterogeneous lung disease, the application of positive airway pressure can modulate and redistribute blood flow away from ventilated regions and toward unventilated regions of the lung by directly increasing the pulmonary vascular resistance of lung segments exposed to elevated airway pressure, that is, segments not protected by consolidation or airway obstruction.44
Evidence suggests that basal release of NO and the subsequent increase in smooth muscle cell concentrations of cGMP partly mediate the low resting pulmonary vascular resistance of the newborn.28,71 Other vasoactive substances—including histamine, 5-hydroxytryptamine, bradykinin, and metabolites of arachidonic acid by the cyclooxygenase and lipoxygenase pathways—have been implicated in mediating postnatal pulmonary vascular tone. However, their precise roles are not well elucidated.
Two of the most important factors affecting pulmonary vascular resistance in the postnatal period are oxygen concentration and pH. Decreasing oxygen tension or pH elicit pulmonary vasoconstriction of the resting pulmonary circulation.72 Alveolar hypoxia constricts pulmonary arterioles, diverting blood flow away from hypoxic lung segments and toward well-oxygenated segments.73 This process enhances ventilation-perfusion matching and is a unique pulmonary vascular response to hypoxia, which is probably greater in the younger animal than in the adult.74 The mechanism of alveolar hypoxic pulmonary vasoconstriction, which remains to be defined, is the subject of several extensive reviews.75 Acidosis potentiates hypoxic pulmonary vasoconstriction, and alkalosis reduces it.72 The exact mechanism of pH-mediated pulmonary vasoactive responses is incompletely understood but appears to be independent of PaCO2.76 Alveolar hyperoxia and alkalosis often are used to relax pulmonary vascular tone because they generally relieve pulmonary vasoconstriction while having little apparent effect on the systemic circulation as a whole. However, detrimental effects of hypocarbia or respiratory alkalosis on cerebral and myocardial blood flow may occur.77
A selective pulmonary vasodilator was long sought for treatment of pulmonary hypertension because (with the exception of oxygen) the response of the pulmonary circulation to humoral vasoactive agents is generally similar to that of the systemic circulation. To date, inhaled NO is the most commonly used agent for selective pulmonary vascular dilation. It is worth noting that its selectivity is not based on a differential effect in the pulmonary and systemic circulations. Rather, when delivered as an inhaled gas, NO is rapidly bound to hemoglobin and inactivated, thus limiting its effects to the pulmonary circulation. Other agents with rapid metabolism (i.e., inhaled prostacyclin) also are being studied via inhalational delivery routes to take advantage of this pharmacologic selectivity.78
Cerebral Circulation
In adults, the brain (which makes up 2% of body mass) receives approximately 14% of the cardiac output and accounts for close to 20% of the body’s O2 consumption. In neonates, the brain receives up to 30% of cardiac output and accounts for 50% or more of total oxygen consumption and up to 98% of produced hepatic glucose consumption. Other organ systems receive a larger percentage of the total cardiac output (e.g., the lung) and utilize greater amounts of oxygen (e.g., skeletal muscle), but the brain is unique in its intolerance for diminished blood flow. In fact, although some favorable outcomes have been reported, severe if not irreversible damage often occurs after just minutes of circulatory arrest under normal conditions.79–82
Blood is supplied to the brain by the carotid and vertebral arteries. The common carotid artery bifurcates, forming the internal carotid artery (ICA) and external carotid artery. The ICA gives rise to its major branches—the posterior communicating, anterior cerebral, middle cerebral, and anterior choroidal arteries—which comprise the anterior circulation. The vertebral arteries give rise to the anterior spinal and posterior inferior cerebellar arteries before merging to form the basilar artery, which in turn gives rise to the anterior inferior cerebellar, superior cerebellar, and posterior cerebral arteries. These vessels comprise the posterior circulation. Venous drainage from the superficial and deep cerebral veins enters the dural sinuses and exits the cranium mostly via the internal jugular vein, although the vertebral veins are important secondary pathways available for venous outflow.83
At rest, CBF is approximately 50 mL/100 g tissue/min.84 Cerebral oxygen consumption is surprisingly high, averaging 3.2 mL/100 g tissue/min. Glucose is the primary energy substrate, although ketones can be utilized during periods of starvation. The brain has no functional capacity to store energy and thus is completely dependent on a steady supply of O2 because up to 92% of its ATP production results from the oxidative metabolism of glucose.79–82
An important feature unique to the cerebral circulation is the presence of a blood-brain barrier (BBB). The vascular endothelium of brain capillaries forms a continuous sheet, with adjacent cells joined by tight junctions. Unlike the endothelium of nonneural capillaries, vascular endothelium does not contain intercellular clefts through which water-soluble particles can traverse, and pinocytosis is markedly diminished. However, the lipid-soluble substances CO2 and O2 can freely diffuse across the endothelium. Metabolically important components such as glucose, lactate, and amino acids depend upon specific carrier proteins to facilitate their diffusion into the brain. Furthermore, the BBB has a biochemical component, with high levels of degradative enzymes that protect the vascular smooth muscle and extracellular fluid from the effects of circulating vasoactive substances, such as catecholamines.85 Thus, as a result of the BBB, the cerebral vasculature responds differently than other vascular beds to humoral stimuli. However, humoral stimuli can significantly alter the vascular tone of large cerebral arteries and can affect blood flow to parts of the brain that lack a complete BBB such as the choroid plexus, the median eminence, and the area postrema.86,87
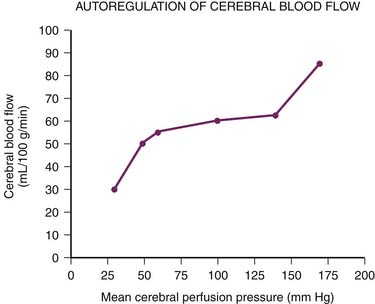
For more than 60 years it has been recognized that CBF remains constant over a wide range of mean systemic arterial pressures88 (Figure 20-11). Constant CBF is maintained in the face of increasing inflow pressures by compensatory vasoconstriction. Conversely, in the setting of low systemic arterial pressures (i.e., low inflow pressures), the cerebral vasculature dilates to maintain steady CBF. At systemic arterial pressures outside the autoregulatory range, further dilation or constriction can no longer maintain blood flow. At high pressures, disruption of the BBB ensues, with subsequent edema and even hemorrhage from ruptured cerebral vessels. At low pressures CBF begins to fall, with continued decreases leading to ischemia and ultimately brain death.80,89–91 Importantly, normal cerebral autoregulation can be impaired in the setting of disease. Traumatic brain injury, subarachnoid hemorrhage, and stroke, for example, all can abolish or impair the normal autoregulatory response.92–95
Although the brain’s ability to autoregulate flow is well established, the mechanisms underlying it are not completely understood.91 A myogenic response appears to be especially important in the setting of raised CPP. Large- and medium-sized cerebral arteries, including the ICA, have been shown to constrict both in vitro and in vivo in response to elevated transmural pressures. While small arteries and arterioles primarily modulate cerebral resistance during normotension, at higher pressures (greater than 110 mm Hg), the large cranial vessels dominate. Thus at high perfusion pressures, smaller more delicate vessels are protected by changes in upstream resistance.91,96
In marked contrast to other vascular beds, neural stimuli have relatively little effect on basal CBF. Cerebral vessels display extensive perivascular innervation, especially by the sympathetic nerves arising from the superior cervical sympathetic ganglia, but the brain is well protected from circulating catecholamines by the BBB. Thus many of the vasoactive agents used in the critical care setting (α- and β-adrenergic agonists) have minimal effects on resting cerebral vascular tone. Mild to moderate electrical stimulation, as well as surgical resection of both the sympathetic and parasympathetic nervous system, does not alter cerebral vascular tone under resting conditions. However, vigorous sympathetic stimulation, as would occur with strenuous exercise or hypertension, does result in vasoconstriction of large- and medium-sized cerebral vessels.82 Thus while a neurogenic mechanism may not mediate cerebral vascular resistance under normal conditions, it does provide protection during times of stress.96–98 Indeed, patients with chronic hypertension have been shown to have a rightward shift of the autoregulatory curve.82,91
With the advent of sophisticated methods of detecting and measuring CBF, such as radioactive xenon, positron emission tomography scanning, and functional magnetic resonance imaging (MRI), intracerebral regional circulatory variations could be mapped and were found to correlate with local central nervous system activation.84,99,100 Blood flow to the occipital cortex increases with visual stimulation.101 Motor activity augments flow to the motor strip and speech enhances flow to Broca’s area.102,103 CBF decreases during deep sleep but returns to normal awake levels during rapid eye movement sleep.104
As in other vascular beds, it appears that CBF is linked or “coupled” to changes in metabolism.105–107 For example, hypothermia decreases the cerebral metabolic rate of oxygen, and therefore CBF, in both animal and human studies.108–111 Seizure activity and fever both increase the cerebral metabolic rate of oxygen and CBF, which explains the deleterious consequences of both conditions for patients with raised ICP.112 The mechanisms underlying this coupling of blood flow and metabolism are still unclear. A number of substances have been shown to affect cerebrovascular tone, including carbon dioxide, oxygen, hydrogen ions, lactic acid, histamine, potassium ions, prostaglandin, ET-1, NO, and adenosine.80,82
Carbon dioxide plays a critical role in the regulation of CBF. In fact, a linear increase in CBF is seen with increasing PaCO2, making CO2 one of the most potent known cerebral vasodilators.113,114 Carbon dioxide exerts its effect via a reduction of the perivascular pH. Whereas arterial H+ cannot cross the BBB, CO2 can easily diffuse into the brain. Carbonic anhydrase facilitates the reaction between CO2 and H2O, forming carbonic acid with subsequent dissociation producing H+ ions. Perivascular acidosis dilates the cerebral vasculature, while alkalosis leads to vasoconstriction.115,116 In this way the cerebral vasculature is distinct in that respiratory acidosis and alkalosis alter tone and CBF, while metabolic acidosis and alkalosis do not.114,117 Interestingly, abnormal CO2 reactivity has been associated with several disease processes, including traumatic brain injury, subarachnoid hemorrhage, stroke, carotid stenosis, and congestive heart failure.92–94 Indeed, abnormal CO2 vasoreactivity has been used as a means to prognosticate in some disease states.94,95
Several studies have demonstrated that the cerebral vasculature adapts in the setting of chronically elevated PaCO2, with changes in the pH of the brain extracellular fluid.118 This finding has obvious implications for the clinician attempting to treat raised ICP with chronic hyperventilation.
Arterial oxygen tension (PaO2) also participates in the regulation of CBF. Arterial hypoxia dilates cerebral vessels at Pao2 below 40 to 50 mm Hg (Figure 20-12).113 The relation between CBF and arterial oxygen content is almost linear, and cerebral oxygen delivery can be maintained unless arterial oxygen content falls below 4 vol%. Hyperoxia does not appear to be a very potent stimulus for vasoconstriction, however.113,119 Hypoxic vasodilation is not limited to the cerebral circulation. Efforts to fully elucidate this phenomenon continue, but it is known that adenosine and both Ca2+-activated and ATP-activated potassium channels are particularly important. Adenosine, which leads to vasodilation through an increase in cyclic adenylate monophosphate, has been found to increase more than fivefold with hypoxia.120–123 Adenosine also is critical in autoregulation, with experimental data revealing a sixfold rise in brain adenosine levels as mean arterial pressure falls from 135 mm Hg to 45 mm Hg.124
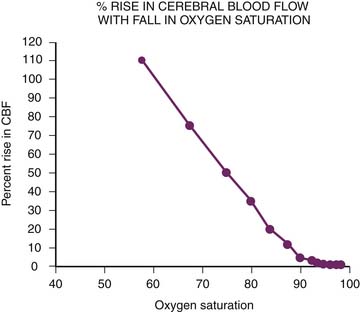
Figure 20–12 Arterial hypoxia dilates cerebral vessels and maintains cerebral oxygen delivery. CBF, Cerebral blood flow.
(Modified from Fuhrman BP: Regional circulation. In Fuhrman BP, Shoemaker WC, editors: Critical care: state of the art, vol 10, Fullerton, CA, 1989, Society of Critical Care Medicine.)





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


