44 Principles of Gas Exchange
 Oxygen Exchange
Oxygen Exchange
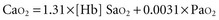
Except in extreme conditions under which Hb is unable to bind O2 (e.g., carbon monoxide intoxication, methemoglobinemia) or under which very severe anemia limits the amount of O2 that can bind to Hb, dissolved O2 accounts for a very small percentage of the total.1 In fact, Hb is such an effective carrier for O2 that the quest to develop an effective blood substitute for clinical use has been only partially successful. Intravascularly delivered products based on stroma-free Hb (an avid oxygen binder) and perfluorocarbon (an efficient dissolver of oxygen) are potentially effective but have encountered problems with stability, toxicity, and cost.2 For the present, blood substitutes must be considered impractical for the clinical setting.
Oxygen Delivery
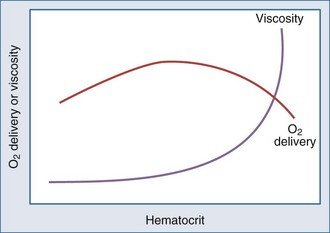
Metabolizing tissues require an adequate supply of oxygen to efficiently produce the energy needed for cellular function. The quantity of oxygen loaded onto the arterial bloodstream per unit time (O2 delivery) is the product of the cardiac output and the oxygen contained within each milliliter of blood. Therefore, a deficiency of either cofactor can be partially offset by a compensatory increase of the other. Conversely, sluggish blood flow, whether caused by low cardiac output or high resistance through the tissues, can limit the O2 actually delivered to the cell. Increased blood viscosity impedes the transit of erythrocytes through the capillary bed, tending to limit oxygen consumption (VO2).3 For this reason, paraproteinemia, extreme leukocytosis, and polycythemia can pose life-threatening challenges to O2 consumption that are independent of any impact on cardiac output.3 Studies performed in animal models demonstrate that hematocrit (Hct), a primary determinant of viscosity, bears a nonlinear relationship to oxygen delivery that varies somewhat with circulating blood volume.4 At low values of Hct, a rising Hb concentration predictably adds to O2 content and delivery. Above a Hct of 30% to 34%, however, it is difficult to demonstrate in critically ill patients an increase in oxygen consumption or an outcome benefit that derive from increases of O2 content arising from further increments of Hb.5 At an Hct of around 55% to 57%, O2 delivery reaches its maximum in normal subjects, falling sharply with each further rise in Hct (Figure 44-1). Above an Hct of approximately 65%, phlebotomy may be required to avert a hemodynamic crisis, as vital tissues may be deprived of delivered oxygen. Viscosity, and therefore tolerance for higher Hct, is partially determined by the circulating blood volume; the polycythemia associated with intravascular volume contraction is much less well tolerated than that of polycythemia vera, a condition in which circulating blood volume is expanded.3 As might be expected, patients with vascular disease are less tolerant to the adverse rheologic effects of high Hct.
At the mitochondrial level, oxygen acts as the terminal acceptor in a chain of organic electron donors known as cytochromes. The PO2 within the mitochondrion needed to sustain this process is very low—estimated to be much less than 1 mm Hg.6 To provide that needed level of mitochondrial oxygen tension, an appropriate oxygen diffusion gradient must be established from the arterial blood, across tissue and cellular boundaries, and into the cellular organelles. At sea level, normal levels of mitochondrial O2 are achieved at a PaO2 of about 95 mm Hg. The actual PO2 within the mitochondrion, however, is affected by many factors other than arterial PO2: tissue metabolic rate, microvascular control, tissue properties, and blood flow. Over time, varying degrees of accommodation to subnormal PaO2 occur by adjustments of the cardiovascular system, Hb concentration, capillary system, and mitochondrial density.7 Although this adaptive phenomenon is commonly observed in patients with chronic lung diseases, the extent to which gradual accommodation to hypoxemia can occur and should be encouraged in patients who are critically ill is a provocative and largely unexplored question.
Oxygen Transfer Across the Lung
Low FIO2 is an important mechanism of hypoxemia occurring at altitude and in fires that occur in confined spaces. Although the relationship is not a strictly linear function, as a rough estimate, inspired O2 declines approximately 15 mm Hg for each 1000 meters of altitude above sea level.8 For practical purposes, however, a reduced concentration of inspired oxygen does not account for hypoxemia that occurs in the setting of critical illness. Hypoventilation alters the alveolar oxygen tension (PAO2) in proportion to the rise of PaCO2 (and PACO2) and becomes an important factor when it occurs during breathing of room air (as in narcotic overdose) or of relatively low inspired concentrations of supplemental oxygen (e.g., via nasal cannulae). The importance of impaired diffusion as a hypoxemic mechanism is sometimes debated, since the transfer of O2 from alveolus to Hb usually requires only a brief time for completion—somewhat less than the normal transit time of the erythrocyte through the capillary.9 Yet under many conditions that are commonly encountered, fewer capillaries are available to accept the cardiac output, so the rate at which blood flows through the lung is accelerated. Simultaneously, diffusion distances are lengthened, and the driving gradient is reduced by disease. For this reason, impaired diffusion is likely to contribute to hypoxemia occurring in the stressed patient with critical illness who receives near-normal FIO2.
Whereas the relationship of FIO2 to PaO2 is more or less linear for the first three oxygen-responsive mechanisms already covered, the response to oxygen supplementation for V/Q imbalance depends on the distribution of abnormal V/Q units contributing to the problem.10 Hypoxemia due to a relatively small number of lung units with very low V/Q characteristics may not respond noticeably to supplemental oxygen unless a very high FIO2 is employed. Conversely, a lung comprised predominantly of lung units with mild V/Q impairment tends to respond in more linear fashion (Figure 44-2). It is also possible to convert very poorly ventilated lung units into airless, unventilated units with inspired gas having a very high FIO2, owing to replacement of unabsorbable nitrogen with diffusible oxygen, leading to the unit’s contraction and eventual collapse as this process continues below the closing volume of the compromised region (absorption atelectasis).11 Unless compensation by hypoxic vasoconstriction is complete, raising FIO2 can paradoxically increase shunt even as it improves O2 transfer in units that remain patent.
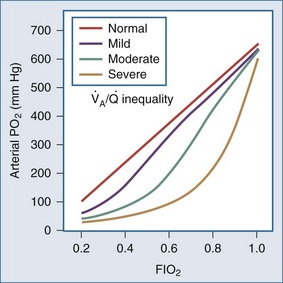
Figure 44-2 Influence of severity of ventilation/perfusion inequality on the FIO2/arterial PO2 relationship.
Given the importance of matching blood flow to ventilation, it is not surprising that several mechanisms have developed to effect pulmonary microvascular regulation. Autonomic control, although less prominent and less precise than in the peripheral vasculature, is important nonetheless. Severe head injury, for example, can cause dysregulation and hypoxemia via this mechanism.12 Local acidosis, such as that existing in poorly ventilated areas, tends to vasoconstrict the pulmonary arterial microvessels. The strength of this reflex, however, pales before that of hypoxic pulmonary vasoconstriction, which for most individuals is a well-developed protection against the consequences of perfusing underventilated areas.13 These mechanisms may be overpowered by pathologic processes or by pharmacologic interventions. For example, local release of inflammatory mediators or use of certain vasoactive drugs (e.g., nitroprusside) may counter these protective reflexes,14 and an abrupt rise of pulmonary artery pressure may overwhelm them.
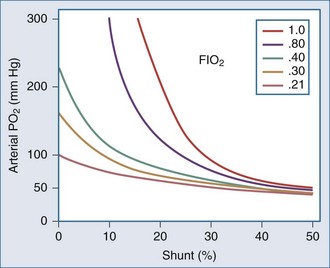
Shunting occurs when systemic venous blood is not brought into close proximity with the inspired gas. Shunt can originate in the heart (e.g., through a patent communication at the atrial or ventricular level). Rarely, direct venous-to-arterial transfer occurs through micro- or macrovascular defects known as pulmonary arteriovenous fistulae. Such communications are encountered in relatively common diseases such as hepatic cirrhosis as well as in other settings, exemplified by the heritable Osler-Weber-Rendu abnormality. Diseases that affect the lung parenchyma are much more common causes of shunt than these cardiovascular disorders. Filling of the airspaces with fluid (e.g., edema) or cellular infiltrate (e.g., pneumonia) prevents effective gas-blood interchange. Inflammatory conditions may inhibit hypoxic vasoconstriction, worsening arterial hypoxemia, as does hypocapnic alkalosis.15 Collapse of lung units may occur on any anatomic scale, resulting in shunt through the affected regions. Causes for collapse vary from compression (e.g., by a pleural effusion), to disease-induced surfactant depletion or inactivation, to airway plugging, such as by retained secretions or a misplaced endotracheal tube. Sustained reversal of atelectasis requires attention to the inciting cause as well as recruitment of the problem area by deep lung expansion. Pure oxygen breathing will not improve hypoxemia due to shunting. Conversely, reduction of FIO2 will not cause shunt-related hypoxemia to worsen and may spare ventilated areas the exposure to potentially toxic concentrations of inspired O2 (Figure 44-3).
Modification of body position may dramatically affect shunting and blood oxygenation when the lungs are injured, especially when disease is asymmetrically distributed. Prone positioning routinely improves oxygen exchange by altering the distribution of transpulmonary pressure as it modifies chest wall compliance and allows the heart to sink to a dependent position that does not compress the lungs. The dorsal lung zones, which are generally the best perfused, tend to reopen when prone. Drainage of secretions and lymphatic efficiency may also improve (Figure 44-4).
 with a fiberoptic Swan-Ganz catheter enable venous desaturation to be detected and monitored.
with a fiberoptic Swan-Ganz catheter enable venous desaturation to be detected and monitored.Venous oxygen saturation is a clinical tool to evaluate the relationship between oxygen uptake and delivery for the whole body. In the absence of pulmonary artery catheter–derived mixed venous oxygen saturation (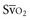 ), the central venous oxygen saturation (ScvO2) is increasingly being used as an imprecise but convenient surrogate measure. Central venous catheters are simpler to insert, less expensive, and associated with fewer complications than pulmonary artery catheters. Blood sampling through central venous catheters allows measurement of ScvO2 or even continuous monitoring if a fiberoptic oximetric catheter is used. The normal range for
), the central venous oxygen saturation (ScvO2) is increasingly being used as an imprecise but convenient surrogate measure. Central venous catheters are simpler to insert, less expensive, and associated with fewer complications than pulmonary artery catheters. Blood sampling through central venous catheters allows measurement of ScvO2 or even continuous monitoring if a fiberoptic oximetric catheter is used. The normal range for 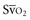 is 68% to 77%, and ScvO2 is considered to be approximately 5% above these values.16,17
is 68% to 77%, and ScvO2 is considered to be approximately 5% above these values.16,17
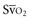 (or Scv
(or Scv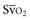 are linked by a simple equation:
are linked by a simple equation: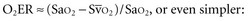
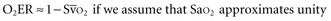
In human studies, dysoxia is usually present when 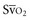 falls below 45%. Tissue oxygen privation may occur at higher levels of
falls below 45%. Tissue oxygen privation may occur at higher levels of 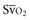 when oxygen extraction is impaired. Ideally, efforts to boost cardiac output (by intravenous fluids or inotropes), Hb, and/or arterial oxygen saturation return
when oxygen extraction is impaired. Ideally, efforts to boost cardiac output (by intravenous fluids or inotropes), Hb, and/or arterial oxygen saturation return 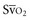 to levels above 65% (or ScvO2 to ≥70%).16,18
to levels above 65% (or ScvO2 to ≥70%).16,18
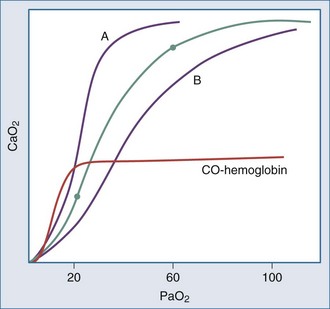
Relationship of PO2 to Blood O2 Content
Even though the oxyhemoglobin dissociation relationship is implicitly used for clinical decision making, there are important nuances (Figure 44-5). Over the clinically relevant range, the oxyhemoglobin dissociation curve is highly nonlinear, so that a drop of a few percentage points in SaO2 over the 95% to 100% interval reflects a much larger change in PaO2 than does a similar decrement that occurs over the 80% to 85% interval. Pulse oximeters record the relative absorption of light by oxyhemoglobin and deoxyhemoglobin. For a fixed value of Hb, the O2 saturation parallels its relative O2 content, but a high saturation guarantees neither its total O2 content nor the adequacy of tissue O2 delivery. For example, a patient may have a “full” SaO2 after inhaling a high concentration of carbon monoxide, and yet directly measuring arterial oxygen content per deciliter of blood (e.g., using a co-oximeter) may demonstrate profound arterial O2 depletion (see Figure 44-5). Moreover, a patient in circulatory shock may maintain a perfectly normal SaO2 despite serious O2 privation. Because cyanide blocks the uptake of oxygen by the tissues, O2 consumption is low in cyanide poisoning, even though arterial and mixed venous saturations are normal or increased. It is occasionally forgotten that arterial oxygen saturation bears no direct relationship to the adequacy of ventilation; a patient breathing a high inspired concentration of oxygen will maintain a nearly normal SaO2 for a brief period in the face of a full respiratory arrest.
< div class='tao-gold-member'>


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


