Chapter 116 Principles of Drug Disposition in the Critically Ill Child
Drug Disposition in Infants and Children
It should come as no surprise that controversy exists regarding drug dosing in pediatric patients. Over the years, a number of dosing rules been developed with the intent that drugs be safely administered to young children. All these rules depend on the standard adult dose with a scale-down factor based on body weight or age. However, distinct differences in pharmacokinetics and pharmacodynamics (Box 116-1) distinguish the pediatric patient from the adult patient. Critical illness may further alter pharmacokinetics and pharmacodynamics in children. These differences must be recognized before providing safe and effective dosing and during the initial selection of the drug itself.
Determinants of Effective Therapy
Effective therapy results when the drug(s) selected for a given condition has both favorable pharmacokinetic and favorable pharmacodynamic properties (see Box 116-1). Moreover, administration to the patient must be individualized based on (1) a realistic clinical endpoint determined before administration; (2) sound knowledge of the quantitative aspects of the disposition of the drug selected; and (3) an understanding of the impact on the patient’s illness of both the dosing regimen to be used and the anticipated therapeutic effect.
Pharmacokinetics
Drug Absorption
Absorption of drugs from the gastrointestinal tract is affected by a number of factors (Box 116-2).1,2 In general, enteral absorption depends on gastric emptying, intestinal surface area and motility, and hepatic first pass. Ontogeny and critical illness may significantly affect these and other patient factors (Figure 116-1 and Box 116-3).
Bioavailability (F) describes the fraction of a dose of drug that reaches the systemic circulation. Bioavailability of a single drug may vary significantly depending on the route of administration. By routes other than intravenous, absorption is a primary determinant of F. In enteral administration, an additional factor influences F. Excluding drugs primarily absorbed by the oral mucosa, drugs administered into the gastrointestinal tract may undergo metabolism by intestinal mucosal cells and/or metabolism and/or biliary excretion when they pass through the liver, before reaching the systemic circulation. This is known as the first-pass effect. With affected drugs, this phenomenon may significantly reduce F. This accounts for the fact that the enteral dose for many drugs is significantly greater than the intravenous dose. The susceptibility of a drug to hepatic first-pass metabolism may influence how the drug is administered; for example, nitroglycerin is given sublingually to circumvent a considerable first-pass effect in this drug.3 Aside from the physicochemical nature of the drug itself, other factors may influence the extent of hepatic first-pass metabolism. Changes in hepatic blood flow may alter this action.4 Age likely further influences the extent of hepatic first pass. As described later in the chapter, maturation of hepatic enzyme systems and transporters appears to occur postnatally.7 Although the data dedicated to the ontogeny of metabolizing enzymes and transporters in the liver are limited, particularly as they relate to drug bioavailability, F probably decreases with age as these systems mature.7
Intramuscular Administration
The parenteral route of drug administration is important when a patient’s disease state precludes oral therapy or when the bioavailability of an oral formulation is poor. The intravenous route for drug delivery is preferred over intramuscular (IM) injection. However, in children with poor intravenous access, IM injection is a viable and effective alternative for the administration of many drugs. Both physicochemical and physiologic factors affect the rate of drug absorption from the IM injection site.8 Lipophilicity of a drug favors rapid diffusion into the capillaries; however, the drug must retain a degree of water solubility at physiologic pH to prevent precipitation at the injection site. For example, the sodium salt of phenytoin is principally an acid and thus is insoluble in the extracellular milieu of skeletal muscle. This explains the poor IM absorption of phenytoin. By contrast, phenobarbital and benzathine penicillin are well absorbed after IM administration. Both of these drugs are weak acids with pKa values close to physiologic pH and are therefore unlikely to precipitate in muscle under most physiologic conditions. By having knowledge of the physicochemical properties of a drug preparation, the clinician can predict, even control to some extent, how the drug is absorbed. While aqueous preparations will undergo rapid absorption, drugs in a solution of oil or other repository vehicles will be absorbed at a slower and more continuous rate.9
Another important factor that influences absorption of drug from an IM injection site is local blood flow, which may be compromised in patients with poor peripheral perfusion.10 Rate and extent of absorption from an IM injection site also are influenced by the total surface area of muscle in contact with the injected solution, similar to the dependency of oral absorption on the total available absorptive area in the intestines.8
A final consideration in IM absorption is muscle activity, which may affect the rate of absorption and therefore affect the peak serum concentration. Sick, immobile infants and children or those chemically paralyzed may show reduced absorption rates after IM drug administration. Use of this route of administration in the ICU may be limited by actual or induced (anticoagulant therapy9) bleeding diatheses in some patients. A list of intramuscularly administered drugs used frequently in the pediatric intensive care unit (PICU) is provided in Box 116-4.
Subcutaneous Absorption
As with absorption of drugs from IM sites, absorption of subcutaneously administered drugs is influenced by local blood flow, as well as by proximal scarring or injury.4 The pattern of absorption varies similarly to that following IM injection, depending on the physicochemical properties of the preparation. Frequently, drugs given by the subcutaneous route undergo slow and sustained absorption.9 As such, the rate of absorption can be regulated by the drug formulation. For example, when drugs are administered in solid pellet form, absorption may occur over weeks to months.9 Absorption can be slowed by the addition of a vasoconstrictor.9 This route of administration is not appropriate for large volumes or for drugs that are irritating to tissues.
Percutaneous Absorption
Percutaneous absorption is inversely related to the thickness of the stratum corneum and directly related to skin hydration.11 The stratum corneum is generally assumed, but not proven, to be thinner in children than in adults. The integument of the full-term neonate possesses intact barrier function.12 This is not assured in the case of the premature infant.6 Another important factor dictating percutaneous absorption is the surface area/body weight ratio, which is much larger in the full-term neonate than in an adult. Theoretically, if a newborn receives the same percutaneous dose of a compound as an adult, the systemic availability per kilogram body weight is approximately 2.7 times greater in the neonate.
The percutaneous route of drug administration is taking on greater importance in the ICU setting. Historically, the most commonly used cutaneous preparation for systemic therapy is nitroglycerin.13 More recently, advances in the technology associated with drug delivery systems have resulted in the common use of clonidine as a percutaneous preparation for treatment of hypertension and narcotic withdrawal. In addition, a number of narcotics exist as cutaneous preparations so that essentially “continuous infusions” of these drugs can be safely administered outside the ICU.14 Finally, even drugs such as nitroglycerin ointment are finding potential new uses in the PICU, for example, treatment of the distal ischemia associated with purpuric injuries.
Rectal Absorption
Rectal administration of drugs is of potential therapeutic importance if a patient cannot take an agent orally and intravenous access for drug administration is impracticable. Because of the routes of the respective venous drainage systems, drugs administered into the superior aspect of the rectum are susceptible to hepatic first pass, whereas drugs administered lower into the rectum initially bypass the liver.15 This may be an advantage for drugs such as lidocaine or propranolol that demonstrate a significant hepatic first-pass effect. The predominant mechanism for drug absorption from the rectum probably is similar to that observed in the upper gastrointestinal tract (i.e., passive diffusion). Theoretically, the physicochemical and host factors discussed earlier with respect to oral drug absorption also influence rectal drug absorption. In general, absorption from aqueous or alcoholic solutions is more rapid than from suppositories.
Lipophilic drugs with pKa between 7 and 8, such as barbiturates and benzodiazepines, seem to be ideally suited for rectal administration because they exist mostly in unionized form and readily cross cell membranes. Rectal use of drugs such as thiopental and diazepam may be effective when intravenous access is a problem and rapid induction of anesthesia is desired or when a child is convulsing. Dulac et al.16 showed that rectal administration of 0.25 to 0.5 mg/kg of a diazepam solution to children aged 2 weeks to 11 years produced serum concentrations comparable with those observed after intravenous administration. Additionally, peak serum concentrations occurred within minutes of administration. Potentially effective anticonvulsant serum concentrations were maintained for 1 to 3 hours in most of the study patients. Knudsen17 demonstrated similar results in 20 children aged 1 to 2 years and further ascertained the clinical efficacy of rectal diazepam in preventing recurrent febrile seizures. In a similar fashion, Burckart et al.18 reported rapid and effective sedation after rectal administration of thiopental suspension to 36 infants and children undergoing computed tomographic scanning.
Drug Distribution
Knowledge of drug distribution is important for selecting the appropriate drug and dose to be administered. Distribution of most drugs in the body is influenced by a variety of age-dependent factors, including protein binding, body compartment sizes, hemodynamic factors such as cardiac output and regional blood flow, and membrane permeability.19,20 Any of these factors also can be altered by disease. This section briefly reviews the pharmacokinetic description of distribution and the effect of ontogeny and critical illness on several factors that determine drug distribution.
The primary pharmacokinetic parameter representative of drug distribution is the volume of distribution (Vd). Vd reflects the apparent space within the body available to contain drug and relates the amount of drug in the body to its concentration in a biologic fluid, usually blood or plasma.9 Vd varies among drugs, based on the drug’s extent of protein and tissue binding and its partition coefficient in fat.9 Additionally, for any given drug, Vd varies among patients because of differences in protein stores or binding and body composition as a result of age or illness. For example, in neonates and infants, a relatively increased extracellular fluid volume, decreased protein binding, and increased brain and liver size all contribute to increased weight-normalized Vd for most drugs.21 An understanding of the factors influencing Vd is of paramount importance to the clinician caring for critically ill children. Vd relates the administered dose of drug to its plasma or blood concentration, which determines therapeutic effects, both favorable and adverse. Alterations in Vd produce reciprocal changes in drug concentration.21 Familiarity with the concept of Vd and the ontogenic and other factors influencing its variation will assist the intensive care physician in understanding why a standard drug dose might be inappropriate for a given patient.
Developmental Aspects of Protein Binding
Plasma protein binding of drugs depends on the concentration of binding proteins available, the affinity constant of the drug for the protein(s), the number of available binding sites, and the presence of pathophysiologic conditions or endogenous compounds that may alter drug-protein interaction.22 The affinity of albumin for acidic drugs increases, as do total plasma protein levels, from birth to early infancy.23 These values do not reach normal adult levels until age 10 to 12 months. In addition, although plasma albumin may reach adult levels shortly after birth, the neonatal albumin level in blood is directly proportional to gestational age, reflecting placental transport and fetal synthesis.24 Binding of basic drugs by α1-acid glycoprotein also is affected.21,25 Studies in cord blood suggest that decreased levels of α1-acid glycoprotein cause this decreased binding.25 Some of the drugs that have exhibited decreased protein binding in the infant include diazepam, furosemide, propranolol, thiopental, phenytoin, and some antibiotics.21 The comparative binding of some of these drugs in neonates and adults is given in Table 116-1. The impact of decreased binding of many of these drugs on efficacy has not been determined.21 For drugs that are highly protein-bound and subject to therapeutic monitoring (e.g., phenytoin), however, any decrease in protein binding may result in a lower measured total drug concentration.21 In such cases, monitoring of free drug concentration (if possible) will prove more clinically relevant.
Table 116–1 Comparative Protein Binding of Some Representative Drugs
| Drug | % Bound | |
|---|---|---|
| Newborn | Adult | |
| Acetaminophen | 37 | 48 |
| Ampicillin | 10 | 18 |
| Diazepam | 84 | 99 |
| Lidocaine | 20 | 70 |
| Morphine | 46 | 66 |
| Phenobarbital | 32 | 51 |
| Phenytoin | 80 | 90 |
| Propranolol | 60 | 93 |
| Theophylline | 36 | 56 |
Data from Kurz H, Mauser-Ganshom A, Stickel HH: Differences in the binding of drugs to plasma proteins from newborn and adult man, Eur J Clin Pharmacol 11:463, 1977.
Developmental Aspects of Fluid Compartment Sizes
Alterations in body water compartment sizes affect the volume of distribution of a drug. Age-dependent changes in the various fluid compartments (Table 116-2) were reviewed in detail by Cheek et al.27 and Friis-Hansen.28 Regardless of age, critical illness and related therapies may alter total body water and other fluid compartment volumes.
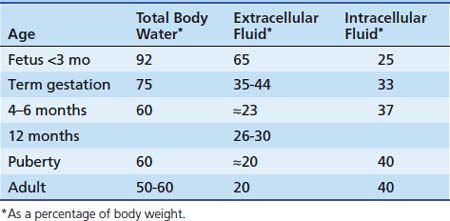
At 40 weeks of gestation, measurements of extracellular fluid volume have ranged from 350 to 440 mL/kg body weight. Cassady29 demonstrated that the extracellular fluid volume of newborn infants correlated more closely with body weight than with gestational age. By age 1 year, extracellular fluid volume decreases to approximately 26% to 30% of body weight. After the first year, it decreases slowly and gradually approaches the adult value of 20% body weight by puberty. Intracellular fluid volume increases from 25% of body weight in the young fetus to 33% at birth to approximately 37% of body weight at age 4 months. Except for a sudden increase during early childhood, the intracellular fluid volume remains relatively constant thereafter, approximating 40% of body weight. The clinical relevance of this gradual reduction in the size of extracellular body water compartments with age cannot be overemphasized. To achieve comparable plasma and tissue concentrations of drugs distributing into extracellular fluid, higher doses per kilogram body weight must be given to infants and children than to adults.30
Developmental Aspects of Body Composition
The percentage of body weight composed of adipose tissue approximately doubles over the first year of life.21 Additionally, the liver and brain account for a higher percentage of body weight in the neonate than in the adult.6,21 All of these factors may lead to significant differences in weight-normalized Vd between infants and adults, depending on the drug. Differences in the amount of adipose tissue may alter clearance of some drugs.
Critical Illness and Drug Distribution
Because of impaired production or increased losses, respectively, conditions such as liver disease and nephrotic syndrome reduce circulating albumin concentrations, resulting in an increase in apparent Vd. Diseases that induce an acute phase reaction (e.g., malignancy, myocardial infarction, inflammatory bowel disease) may provoke increased binding of basic drugs because of increased levels of α1-acid glycoprotein.9 Additionally, accumulation of extravascular fluid collections (e.g., pleural effusions and ascites) results in the development of a reservoir for drugs that are distributed into the total body water.31
Drug Clearance
Clearance reflects the removal of drug from the body. Clearance occurs by two processes: biotransformation (i.e., metabolism) and excretion. Metabolism occurs primarily in the liver. Excretion is predominately facilitated by the kidney, although excretion also can occur via exhalation, saliva, sweat, or the gastrointestinal tract.6,9 Redistribution of drug can contribute to total clearance if the reference compartment is blood or plasma, as is often the case.4 When reflected in the blood or plasma, clearance quantitatively describes the volume of blood or plasma from which all drug is removed per unit of time.4,6 Clearance is an important pharmacokinetic parameter to consider when the goal is maintenance of steady-state drug concentration, which correlates with therapeutic efficacy. Once steady state is achieved, clearance of the drug determines the quantity of drug that must be administered in order to maintain that concentration (i.e., Drug out = Drug in).4,7 The clinically relevant pharmacokinetic concepts related to clearance are discussed later in this chapter. The ontogeny of systemic clearance mechanisms probably accounts for a significant portion of the difference in pharmacologic response between infants and adults.7 These developmental factors also produce variability among pediatric patients. Critical illness may have an additional profound impact on clearance mechanisms.
Hepatic xenobiotic metabolism assumes an extremely important role in determining the pharmacokinetic and pharmacodynamic properties of many drugs. Clearance of a drug (Cl) by an individual organ depends on blood flow to the organ (Q) and the organ’s extraction ratio (E) and can be described as follows32: Cl = Q × E, where E is the ratio of the arteriovenous concentration difference divided by the arterial concentration (extraction) as expressed by E = (Ca − Cv)/Ca,where Ca and Cv are the arterial and venous concentrations, respectively. Organ clearance concepts are best described for the liver and kidneys. Hepatic clearance depends on hepatic blood flow, plasma free drug concentration, cellular uptake, hepatic metabolism, and biliary excretion. The hepatic clearance of a drug can be expressed by the following equation:
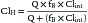
where ClH is hepatic clearance, Q is hepatic blood flow; fB is fraction of free drug; and Clint is intrinsic clearance, which is a measure of hepatocellular metabolism. Drugs that are primarily cleared by the liver can be classified as flow limited or capacity limited.33 If a drug displays a high Clint and E, then doubling the Clint will have little effect on ClH, whereas a change in blood flow will produce a proportional change in ClH. In other words, for drugs that are highly extracted (>80%) and metabolized by the liver, ClH reflects the amount and rate of drug delivered to the liver.34 The considerable declines in hepatic blood flow and oxygen delivery that occur immediately following birth do not appear to translate to significantly reduced clearance of flow-limited drug in the newborn compared with the adult.7 Drugs with high extraction ratios are subjected to the first-pass effect when administered enterally.
At every level, from the ontogenetic changes in hepatic blood and portal oxygen tension to the developmental alterations in protein binding and xenobiotic metabolizing enzyme activities, there is the potential for age to affect the processes associated with hepatic clearance. Very little investigative effort has been expended to elucidate these effects; however, some of the important available data are discussed further. For detailed reviews of this data, the reader is referred to articles by Alcorn and McNamara7 and Leeder and Kearns.35
Biotransformation: Phase I Reactions
The biotransformation of endogenous and exogenous substances occurs primarily in the liver, although the adrenal gland, placenta, kidney, gut, and skin also are capable of metabolizing compounds. Once a drug enters the hepatocyte, it may be transformed by one or more enzymatic reactions. These pathways, or phase I reactions, include oxidation, reduction, hydrolysis, and hydroxylation.36,37 In general, these reactions are responsible for transforming compounds into more polar, less lipid-soluble molecules that are more rapidly eliminated by the kidney, biliary system, or lung. However, parent compounds may be transformed into pharmacologically active intermediates, such as theophylline to caffeine, or into toxic metabolites, as occurs with oxidation of acetaminophen. In addition, pharmacologically inactive parent compounds (prodrugs) may be converted to active moieties, as occurs with hydrolysis of chloramphenicol succinate to chloramphenicol. The ontogeny of human enzyme systems differs dramatically from most animal species, especially for oxidation and glucuronidation pathways.38 Therefore extrapolating data for enzyme system maturation from animals to humans is difficult. Of the enzyme systems capable of metabolizing drugs, the hepatic cytochrome P450 (CYP) mixed-function oxidase system has been studied in greatest detail. It is responsible for most of the phase I reactions catalyzed in the human liver.
Yaffe et al.39 first demonstrated drug-oxidizing enzymes in the human fetal liver. During fetal life, these enzymes are present at 30% to 60% of adult activity in vitro.40 Following birth, total CYP levels increase, approaching adult range by age 1 year.7 Activity of all CYP enzymes is generally thought to be considerably lower in children, particularly neonates and infants, than in adults. In truth, the developmental aspects of expression and function vary among CYP enzyme families and isoforms. Although data delineating these variations are limited, particularly for specific families or isoforms, some insight into the ontogeny of select enzymes has been gleaned from immunochemical studies in hepatic microsomes and tissue, pharmacokinetic studies of known enzyme substrates, and studies evaluating the biotransformation of pharmacologic “probes” (e.g., carbamazepine in the case of CYP3A4). The ontogeny and the drugs affected by some of the important CYP isoforms are outlined in Table 116-3. A limited discussion of the most clinically relevant isoforms in pediatric patients follows.
Table 116–3 Ontogeny of Select Hepatic Enzymes
| Enzyme | Representative Substrates | Developmental Evolution |
|---|---|---|
| PHASE I | ||
| CYP1A2 | Acetaminophen, caffeine, theophylline, warfarin | Negligible in fetal liver. Adult levels of activity by approximately age 4 months. Activity exceeds that in adults after age 1 year; gradually declines to adult levels by end of puberty. |
| CYP2C9 | Diazepam, phenytoin, NSAIDs, tolbutamide, S-warfarin | Undetectable to low in fetal liver. Adult levels of activity by age 1–6 months. Activity exceeds that in adults from age 3–10 years; gradually declines to adult levels by conclusion of puberty. |
| CYP2D6 | Numerous, including captopril, codeine, dextromethorphan, haloperidol, metoprolol, propranolol, ondansetron, tricyclics | Undetectable in fetal liver. Expression and activity appear to be stimulated by parturition. Complete maturation may occur by age 1 year, although acquisition of adult activity levels has been reported to occur as late as age 5 years. |
| CYP3A4 | Numerous, including acetaminophen, amiodarone, budesonide, carbamazepine, cyclosporine, erythromycin, lidocaine, nifedipine, tacrolimus, theophylline, verapamil, R-warfarin | Low in fetal liver; replaces CYP3A7 as the predominant isoform following birth. Based on pharmacokinetic and drug disposition studies, activity in children thought to be greater than that in adults. Decline toward adult levels begins at approximately age 4 years. Adult levels reached by end of puberty. |
| PHASE II | ||
| Uridine 5′-diphosphate glucuronyltransferases (UGTs) | Numerous, including acetaminophen, benzodiazepines, bilirubin, chloramphenicol, dextromethorphan, morphine, naloxone, NSAIDs, propofol, thyroxine | Varies by isoform; difficult to characterize individual isoforms because of overlapping substrate specificities. As a group, activity appears to be deficient in the neonate and infant. Variably reported acquisition of adult levels of activity; anywhere from age 2–30 months, depending on the proposed isoforms involved. |
| N-acetyltransferase-2 (NAT2) | Caffeine, clonazepam, hydralazine, ioniazid, procainamide | Low activity in neonates and infants. Movement toward adult phenotypes (≈50% fast and 50% slow acetylators) after nearly 3 months of age. |
| Methyltransferase group (MT) | Catecholamines, captopril, serotonin, spironolactone | S-methylation capacity (TPMT) approximately 50% greater in infants than in adults. Limited evaluating maturation after this point; one Korean study demonstrated adult level activity by age 7–9 years. |
| Sulfotransferase group (ST) | Acetaminophen, bile acids, chloramphenicol, cholesterol, dopamine, polyethylene glycols, salicylates | At least some isoforms well developed in the infant; compensates for deficient glucuronidation of certain substrates (e.g., acetaminophen). |
Substrate listings from Leeder JS, Kearns GL: Pharmacokinetics in pediatrics: implications for practice, Pediatr Clin North Am 44:55, 1997. Additional data related to substrates and all remaining data derived from references cited in the text.
NSAID, Nonsteroidal antiinflammatory drug.
CYP1A2
Cytochrome P450 1A2, the only CYP1A isoform found in human liver, is involved in the biotransformation of many drugs, including the methylxanthines.35 Immunohistochemical studies have suggested that this protein is sparse, if present at all, in fetal liver.41 These studies also demonstrate that levels of CYP1A2 do not appreciably increase until several weeks to months after birth, remaining below adult levels well into childhood. These findings are reflected in studies examining enzyme activity as assessed by biotransformation of theophylline. Nassif et al.42 reported a significant correlation between a decreasing elimination half-life for enterally administered theophylline and increasing age. Decreased elimination was further suggested by a marked difference in dosage requirement, with patients younger than 4 months requiring approximately half the daily dose needed in patients age 8 to 13 months to maintain therapeutic levels. Tateishi et al.43 evaluated biotransformation of intravenous theophylline, quantifying three metabolites (1-methyluric acid [IMU], 3-methylxanthine [3MX], and 1,3-dimethyluric acid [DMU]) in urine. The ratios of metabolites to theophylline in urine increased dramatically from the neonatal period to age 3 years, when they appeared to essentially plateau. However, a greater variation in these ratios was seen among patients older than 3 years. This study also established that the relative production of DMU, a product of reactions catalyzed by other CYP enzymes, including CYP2E1 and CYP3A4, was higher in the youngest patients compared with those older than 3 years. The relative production of the other two metabolites, which result from CYP1A2 activity alone, is similar between the groups. After age 1 year into early childhood, rates of theophylline clearance appear to exceed the rates in adults, prompting the need for an increased dose to maintain therapeutic levels.35





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


