INTRODUCTION AND EPIDEMIOLOGY
Sudden cardiac arrest represents one of the most time-sensitive diseases in the practice of emergency medicine, requiring prompt recognition and rapid delivery of resuscitative care, including high-quality CPR, early defibrillation when appropriate, and appropriate airway management.1 Even with these interventions, aggregate survival to hospital discharge is less than 20% in most communities and hospital systems.2,3 Among survivors, neurologic injury is common (present in up to 50% of survivors) and widely varied, ranging from subtle memory deficits to persistent vegetative state.4,5 This chapter focuses on the pathophysiology of ischemia-reperfusion injury and the provision of targeted temperature management, also called therapeutic hypothermia.6
PATHOPHYSIOLOGY OF ISCHEMIA-REPERFUSION INJURY
The brain is exquisitely sensitive to ischemia, such that disruption of blood flow for several minutes is sufficient to initiate a set of injury mechanisms that may lead to irreversible disabilities.7,8 The complete loss of blood flow, followed by the abrupt return of spontaneous circulation (ROSC), rapidly leads to a complex pathophysiologic process known as ischemia-reperfusion injury, also known specifically in the sudden cardiac arrest setting as postresuscitation syndrome.8,9
When blood flow is abruptly stopped and then restored, a number of overlapping mechanisms lead to clinical injury (Figure 26-1).
FIGURE 26-1.
Schematic of postarrest pathophysiology, also called ischemia-reperfusion injury. The concepts shown here represent processes that occur at the subcellular, cellular, tissue, and organismal levels. The kinetics of these processes following cardiac arrest and the extent to which they affect clinical outcomes are still unclear.
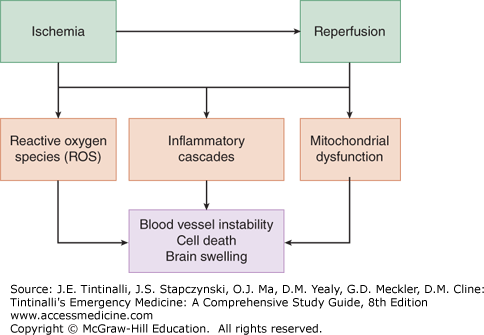
At the cellular level, mitochondrial integrity and function become damaged, with release of crucial enzymatic machinery such as cytochrome c and disruption of oxidative phosphorylation.9,10 Mitochondrial injury is implicated in the increased concentration of oxygen free radicals and downstream activation of programmed cell death pathways.10,11 At the humoral level, reperfusion triggers a broad array of immune activation, including increased blood levels of cytokines including interleukin-6 and tumor necrosis factor alpha.12 In addition, aberrant neutrophil and platelet activation can occur. These immune phenomena have led Adrie et al13 to describe the postresuscitation condition as a “sepsis-like syndrome,” in which inflammation plays a crucial role in injury. Immune activation, in turn, can lead to additional production of oxygen free radicals such as superoxide and hydrogen peroxide. These molecules, produced in small quantities during normal cellular function, are usually converted to oxygen and water by the enzymes catalase and superoxide dismutase. However, enzyme systems become overwhelmed with the dramatic increase of free radical species generated during postresuscitation syndrome. Different tissues exhibit varying sensitivity to ischemia-reperfusion processes, with brain tissue and vascular endothelium appearing to be particularly vulnerable.8,9
The biochemical and cellular phenomena described earlier lead to diverse clinical manifestations of injury in the minutes to hours following resuscitation. Inflammatory changes and immune cellular activation can lead to vascular leak and a drop in peripheral vascular resistance, with concomitant loss of fluid from the intravascular compartment and hypotension.14 Cellular injury in the brain, combined with endothelial damage, can trigger cerebral edema, a common and dangerous component of postresuscitation syndrome.15 Cerebral edema and increased intracranial pressure can cause cerebral herniation and serve as the cause of death, often in the initial 72 hours following resuscitation. Myocardial stunning is clinically evident on echocardiography as global hypokinesis and markedly reduced ejection fraction.16,17 Myocardial depression is usually transient and typically resolves over the first few days following resuscitation with appropriate hemodynamic support.17,18 Postresuscitation infection may be related to bacterial translocation into the bloodstream from loss of intestinal tissue integrity.19 Adrenal injury from ischemia may cause adrenal insufficiency as a further cause of clinical deterioration.20
CLINICAL FEATURES AFTER ROSC
Following initial resuscitation, the clinical condition of patients varies considerably. Some patients with prompt recovery after only brief periods of CPR can be awake and responsive; however, most patients are initially comatose or markedly obtunded following resuscitation. Neurologic assessment is highly limited in most cases, as in the course of clinical resuscitation care, patients are intubated and sedated and/or pharmacologically paralyzed. Patients may be hypotensive or hypertensive, depending on factors such as myocardial stunning, degree of vascular leak, and preexisting hypertensive disease. Patients are most frequently tachycardic following resuscitation; bradycardia may reflect underlying myocardial ischemia and damage to the conduction system.
Physical examination is limited in the immediate postresuscitation period as well. Most patients exhibit absent or abnormal papillary responses and absent gag reflex or doll’s eyes responses. Clinical neurologic reflexes are not predictive of cardiac arrest outcomes.21 The bedside neurologic examination should not influence decisions for continued care in the hours following successful resuscitation. Focus the physical examination after resuscitation on identifying clues to determine the possible cause of cardiac arrest, such as the presence or absence of asymmetric lower extremity edema that might suggest thromboembolic disease, absent or decreased lung sounds that might implicate pneumothorax, or jugular venous distension and muted heart sounds that might suggest cardiac tamponade. An ECG should be obtained and if ST segment elevation consistent with an acute myocardial infarction is present, coronary reperfusion with percutaneous coronary intervention is recommended.
DIAGNOSIS OF ANOXIC BRAIN INJURY AFTER ROSC
Neuroprognostication after sudden cardiac arrest is a crucial issue for healthcare providers in the critical care setting but persists as a challenging and inexact process. A variety of radiographic, biochemical, and neurophysiologic tools are available to assess brain injury following sudden cardiac arrest, but most are poorly validated and require additional research to determine clinical utility.
In the ED, perform head CT in survivors as soon as feasible. Head CT can identify subarachnoid hemorrhage21,22 or epidural and subdural hematomas. The degree of cerebral edema can also be assessed by head CT.22,23,24 Because cerebral edema often peaks several days after resuscitation, swelling that is radiographically apparent on initial CT is a worrisome sign. Brain MRI is generally neither practical nor useful during initial ED management.25
Marked elevations of nerve-specific enolase or S100 calcium-binding protein B are often associated with a poor neurologic prognosis,26 and a nerve-specific enolase level >33 micrograms/L has been used as a cut-off with very high sensitivity for severe brain injury and poor prognosis. The biomarker glial fibrillary acidic protein is under current investigation.27 At present, biomarker measurement is not part of standard emergency medicine practice.
Many protocols for postarrest care involve the early institution of continuous electroencephalogram monitoring to assess for electrical convulsive activity, which is present in up to 25% of patients after resuscitation.28 The presence of seizures generally augurs a poor prognosis, but not all patients with electrical seizure activity sustain severe long-term injury. Bispectral index monitoring, a simple approach to electrical brain monitoring relative to electroencephalography, can be useful to predict neurologic outcome.29,30 Measurement of somatosensory evoked potentials, a bedside test of neuronal connectivity and function, can be performed several days following cardiac arrest and may also be useful to predict outcome.31
TARGETED TEMPERATURE MANAGEMENT (OR THERAPEUTIC HYPOTHERMIA)
The key ED intervention for postresuscitation syndrome is the prompt delivery of systemic cooling therapy, also called therapeutic hypothermia or targeted temperature management. Cooling of resuscitated adult cardiac arrest patients to a core body temperature of 32 to 36°C (89.6 to 96.8°F) for 24 hours following arrest and ROSC can dramatically improve survival and neurologic outcomes.32,33,34 The ideal temperature is not clear. Neurologic outcome after ROSC can be assessed by the Cerebral Performance Score (Table 26-1),35 where a score of 4 or 5 predicts poor quality of life with a sensitivity of 55.6% (95% confidence interval 42-67%) and specificity of 96.8% (95% confidence interval 94-98%). A European study33 demonstrated that therapeutic hypothermia instituted for 24 hours following cardiac arrest and ROSC resulted in a Cerebral Performance Score of 1 or 2 in 55% of hypothermia-treated patients, whereas only 39% of patients in the control group achieved a score of 1 or 2. Six-month mortality was 41% in the hypothermia group and 55% in controls. The trial only enrolled out-of-hospital cardiac arrest patients with ventricular fibrillation/ventricular tachycardia (VF/VT) as an initial rhythm; no definitive trial has been performed for patients with either pulseless electrical activity (PEA) or asystole as the initial arrest rhythm. However, a body of clinical evidence suggests that therapeutic hypothermia also improves outcomes for patients with PEA or asystole.36,37,38,39
| Score | Description |
|---|---|
| 1 | Awake and alert; can work |
| 2 | Awake and alert; cannot work but can perform independent daily activities |
| 3 | Conscious but dependent on others |
| 4 | Coma or vegetative state |
| 5 | Brain death |
Patients who are awake and appropriately alert following ROSC are excluded from consideration (Table 26-2
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


