Fig. 33.1
Normal endothelium with endothelium-derived vasoactive substances. Sheer stress leads to a release of nitric oxide and other vasoactive molecules. [NO nitric oxide, ACE angiotensin-converting enzyme, Ach acetylcholine, AI angiotensin, A11 angiotensin II, ATI angiotensin I receptor, Bk bradykinin]
Portal hypertension , with or without liver disease, causes oxidative stress, and together with the exposure of the endothelium to inflammatory cytokines causes an interruption of the nitric oxide integrity, which results in endothelial dysfunction. This may result in vasoconstriction, microthrombosis, hyperplasia of the vascular muscle layers, and eventual fibrosis. This causes the clinical condition of portopulmonary hypertension. An excess of vasodilatory molecules, nitric oxide, and prostacyclins results in the dilatation of blood vessels, shunt, and aneurysm formation that present as the clinical conditions of hepatopulmonary syndrome.
Portopulmonary Hypertension
Portopulmonary hypertension (POPH) is characterized by an increased pulmonary artery pressure caused by an increase in pulmonary vascular resistance that is the result of portal hypertension usually associated with liver disease [3]. The increase in pulmonary artery resistance involves the presence of an excess of endothelin-1, and other vasoconstrictors such as vasoactive intestinal peptide (Fig. 33.2) [4].
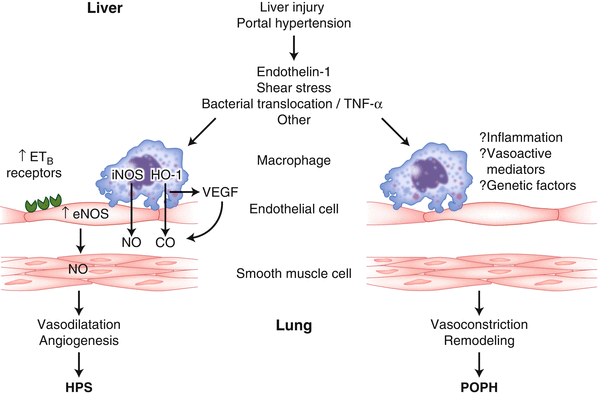
Fig. 33.2
Pathophysiology of hepatopulmonary syndrome (HPS, microvascular dilatation, and angiogenesis) and portopulmonary hypertension (POPH, vasoconstriction, and remodeling in resistance vessels)
There is also development of smooth muscle hyperplasia, hypertrophy, plexogenic arteriopathy, and microthrombi that may be found. Eventually some areas of the microvasculature will progress to fibrosis. All these pathological changes result in an increase in pulmonary vascular resistance that may be reversible by vasodilatation and later remodeling, but areas of fibrosis will result in a fixed defect (Fig. 33.3) [5].
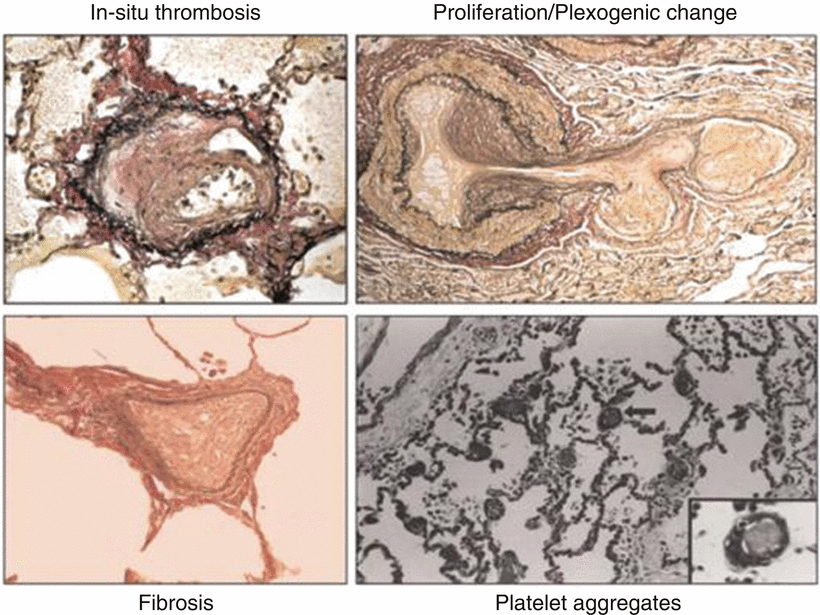
Fig. 33.3
Pulmonary arteriole pathological changes causing an increased resistance to blood flow resulting in portopulmonary hypertension . These images demonstrate intimal thickening, cellular proliferation, fibrosis, and intralumenal microemboli
Portopulmonary hypertension was initially described in 1951 [6]. It is defined as pulmonary arterial hypertension associated with portal hypertension. The portal hypertension is usually associated with liver disease but not always. The diagnosis is made from the hemodynamic data obtained from a right heart catheterization (RHC) . A mean pulmonary artery pressure (MPAP) greater than 25 mmHg at rest and 30 mmHg with exercise, and an elevated pulmonary vascular resistance (PVR) greater than 240 dyn s/cm5, with a transpulmonary gradient greater than 12 mmHg, is pathognomonic of POPH [3, 5]. In many definitions a pulmonary capillary wedge pressure of less than 15 mmHg is included but in patients with severe liver disease this number may be elevated by a very increased cardiac output and volume overload. Therefore, the vascular resistance must be measured by RHC to confirm POPH. See Table 33.1 for a summary of diagnostic criteria for portopulmonary hypertension.
Table 33.1
Diagnostic criteria for portopulmonary hypertension
1. Presence of portal hypertension |
2. Mean pulmonary artery pressure >25 mmHg |
3. Pulmonary vascular resistance >240 dyn s/cm5 |
4. Transpulmonary gradient >12 mmHg |
The incidence of POPH in patients presenting for liver transplantation has been reported to be between 5 and 8.5 % [7–9]. The incidence of pulmonary hypertension in liver candidates is close to 20 % but this is caused by high cardiac output, volume overload, or cirrhotic cardiomyopathy. Mean pulmonary artery pressures of 40–45 mmHg may be found as a result of increased cardiac output, pulmonary venous hypertension, and congestion but on RHC the PVR is found to be normal and the PCWP may be elevated [5]. The resistance across the pulmonary vasculature is not elevated in these patients and therefore it is not POPH [3]. Table 33.2 provides case presentations of pulmonary hypertension in liver transplant recipients as shown by Krowka [5]. Patients #3 and #4 are the only ones with true POPH as they have the elevated pulmonary vascular resistance. Potential causes of pulmonary hypertension are shown in Fig. 33.4 [10].
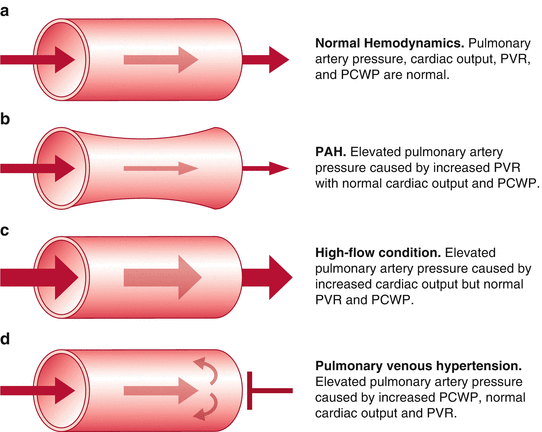
Table 33.2
Pulmonary hypertension presentations in liver transplant recipients
Pt #4 | |||||
|---|---|---|---|---|---|
Pt #1 | Pt #2 | Pt #3 | Before treatment | After treatment | |
RVSP (echo) (mmHg) | 69 | 66 | 99 | 70 | 50 |
MPAP (mmHg) | 33 | 36 | 63 | 50 | 38 |
PCWP (mmHg) | 7 | 25 | 19 | 15 | 15 |
CO (L/min) | 11.9 | 9.3 | 6.1 | 6.3 | 9.3 |
PVR (dyn s/cm5) | 175 | 95 | 577 | 444 | 197 |
Fig. 33.4
Potential causes of an elevated mean pulmonary artery pressure in the patient with liver cirrhosis
Diagnosis of Portopulmonary Hypertension
The clinical presentation of POPH is increasing fatigue, dyspnea on exertion, syncope, and occasional chest pain, and sudden death. In two series of patients with POPH the 4-year and 5-year survivals were 4 % and 14 %, respectively [11, 12]. The most common physical findings are an accentuated pulmonary component of the second heart sound and a systolic murmur. Therefore, on routine clinical assessment these patients are difficult to diagnose, although signs of right ventricular failure may be present. The electrocardiogram may reveal right heart strain. The chest X-ray may show right heart enlargement and failure with dilatation of the pulmonary arteries.
POPH may be precipitated by the increase in cardiac output that may follow a transjugular intrahepatic shunt formation (TIPS) [13].
The most important screening tool is the transthoracic echocardiograph. All patients presenting for liver transplantation should be screened for POPH by transthoracic echocardiography . The right ventricular systolic pressure (RVSP) is estimated based on the velocity of tricuspid regurgitation (TR) using the modified Bernoulli equation RVSP mm Hg = 4 × (TR m/s)2 + right atrial pressure. The tricuspid regurgitant jet flow may not be present in all patients negating this diagnostic tool. In this case a careful assessment of right ventricular function should be made, preferably by transesophageal echocardiography and an estimation of PVR made. One test reporting a sensitivity and negative predictive value of 100 % utilizes the ratio of peak tricuspid regurgitant velocity (TRV) to right ventricular outflow tract velocity time (VTIRVOT) [14]. To confirm the diagnosis an RHC should be performed to clearly characterize the pulmonary hemodynamics.
The Right Ventricle
Assessing right ventricular performance still remains a challenge. The right ventricle (RV) is a complex structure that cannot be approximated by a simple geometric form. It functions in a low-impedance system; therefore it is sensitive to pressure overload. Along with contractility and loading conditions, ventricular interactions play an important part in right ventricular function and failure. Right ventricular dysfunction or failure may result in liver graft congestion and failure and may result in total loss of the newly implanted liver graft and also the recipient. Therefore careful evaluation of the right heart must be made in the pretransplant workup of these patients and a careful assessment of the severity of the POPH must also be made. Most institutions will make an RSVP of >50 mmHg a necessity for an RHC. However, it is not the absolute number of the RVSP or the MPAP that should be the trigger but the function of the RV must be included in this decision. Careful examination by TEE must be made for systolic and diastolic dysfunction of the RV. The RV faced with an increasing pressure overload adapts through hypertrophy and dilatation but eventually will fail. The diagnostic features of RV dysfunction are an E/A ratio <1, a prolonged deceleration time >200 ms, a prolonged isovolumetric relaxation time >80 ms, enlarged right chambers, abnormal pattern of contractility, and a prolonged ratio of pre-ejection period to LV ejection time >0.44 s [15]. Figure 33.5 shows transthoracic echocardiographic images of a patient with portopulmonary hypertension [16].
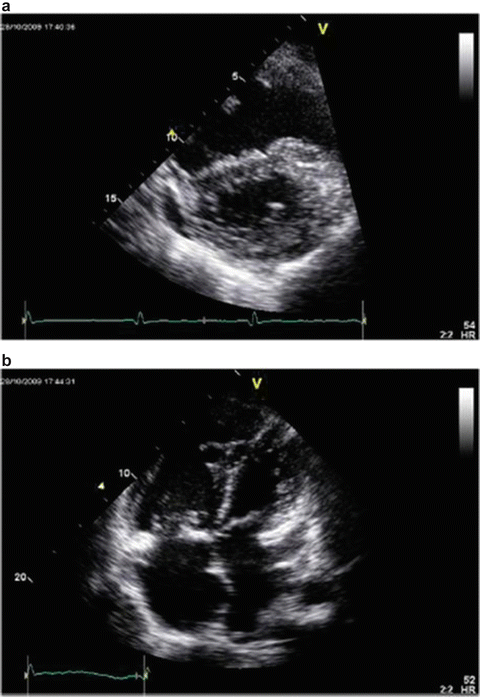
Fig. 33.5
Transthoracic echocardiographic images of a patient with portopulmonary hypertension. Note the D-shaped left ventricle in the short-axis view (a) and a severely dilated right ventricle in the four-chamber view (b)
The typical patient with liver cirrhosis has a hyperdynamic circulation with a low systemic vascular resistance that may mask a significant cirrhotic cardiomyopathy, which can lead to a false sense of security when managing these patients. Most patients with liver cirrhosis will have some manifestations of a cardiomyopathy even if it is just a prolonged QT interval on the electrocardiogram or a downregulation of the beta receptors [17]. This must be taken into consideration when assessing the right heart function in a patient with POPH.
Implications for Liver Transplantation
The key questions that need to be answered are the following: (1) Is it safe for patient and graft to transplant with POPH? (2) Will the POPH resolve after liver transplantation?
Is It Safe to Transplant with POPH?
The data available would suggest that a patient with an MPAP of 25–35 mmHg can safely undergo liver transplantation. Once the MPAP increases above 35 mmHg the mortality increases significantly both for transplantation and on the waiting list for transplantation [18, 19]. A review of the right heart and pulmonary hemodynamics should be made just prior to transplantation to be sure that significant progression of POPH has not occurred since the last evaluation [20]. However, the key to the success of the transplant is the function of the right heart and not the value of the MPAP or PVR. A patient with an MPAP of 30 mmHg with an elevated PVR and poor RV function is at higher risk than the patient with an MPAP of 40 mmHg and elevated PVR but good functioning RV. These patients with right heart dysfunction should be deferred from surgery and have pulmonary vasodilator therapy initiated, and then reevaluated later to assess right heart improvement. Table 33.3 outlines an assessment screening and action plan for patients with portopulmonary hypertension.
Table 33.3
Assessment screen and action plan for portopulmonary hypertension
1. All liver transplant candidates screened with TTE: RVSP > 50 mmHg or RV function questionable; RHC. If MPAP > 25 mmHg and PVR > 240 dyn s/cm5 then TEE to assess right heart function. |
2. MPAP 25–35 mmHg PVR > 240 dyn s/cm5: Good right heart function start pulmonary vasodilator therapy, place on transplant list, and reassess every 6 months. |
3. MPAP 35–40 mmHg PVR > 240 dyn s/cm5: If RV function poor defer transplant and start pulmonary vasodilator therapy and reassess in 6 months. If RV function good then stress RV with dobutamine and fluid challenge; if still good then place on transplant list and start pulmonary vasodilator therapy. |
4. MPAP > 40 mmHg and PVR > 240 dyn s/cm5: Defer transplant and start on vasodilator therapy and reassess in 6 months. |
Does Portopulmonary Hypertension Resolve After Liver Transplantation?
It might seem impossible for patients in whom the intrapulmonary pathology has progressed to fibrosis to have reversal of primary pulmonary hypertension after liver transplantation or with vasodilator therapy. However, there is good evidence that patients who have responded to pulmonary vasodilators will over months after a successful transplant resolve their pulmonary hypertension [21–23].
Intraoperative Management of the Patient with POPH
Those patients that have been assessed to have good RV function and proceed to transplantation still have the potential rigors of a major procedure to undergo and the potential to withstand a 300 % increase in cardiac output after liver graft reperfusion (Fig. 33.6) [20].
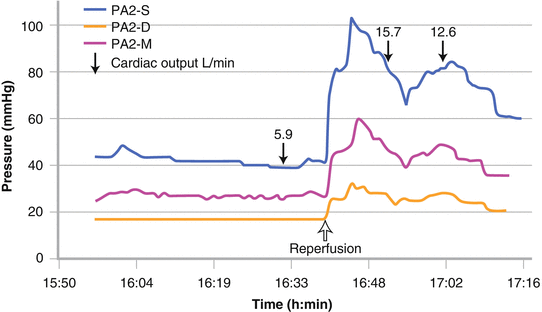
Fig. 33.6
A threefold increase in cardiac output at the time of reperfusion resulting in severe pulmonary hypertension
There is no reliable way to protect the RV and the liver graft if this scenario occurs. Inhaled nitric oxide may be effective in some patients in reversing or moderating this acute rise in pulmonary artery pressure [24]. Consideration for extracorporeal right heart bypass should be given to assist in unloading the RV [25]. Right ventricular assist devices are unlikely to be successful when the RV failure is the result of afterload resistance. Pumping blood into the pulmonary artery will result in increasing pulmonary artery pressure and lung injury [26].
Pulmonary Vasodilator Therapy
Initially the prostacyclins were administered to reduce PVR. However, these drugs had to be given by long-term intravenous therapy (epoprostenol) or by inhalation (inhaled iloprost) [21–23]. Then oral preparations of phosphodiesterase inhibitors, namely sildenafil, became available with promising results [27, 28]. Now two newer drugs, bosentan and ambrisentan, that are endothelin receptor antagonists have been shown to be effective in selected patients [29, 30]. Despite these therapies there are case reports of patients wherein the pulmonary hypertension has progressed after a successful liver transplantation [31]. Perhaps these patients were misdiagnosed with POPH and really had primary pulmonary hypertension in the face of portal hypertension and liver disease.
MELD Exception
Candidates with POPH will be eligible for an MELD exception. Diagnosis should include initial MPAP and PVR levels, documentation of treatment, and post-treatment MPAP < 35 mmHg and PVR < 400 dyn s/cm5. Transpulmonary gradient should be required for initial diagnosis to correct for volume overload [32].
Hepatopulmonary Syndrome
Definition, Incidence, and Clinical Features
In 1884 Flückiger [33] described a female patient with cyanosis finger clubbing and liver cirrhosis. The association between cyanosis and liver disease continued to be observed and a new syndrome termed the hepatopulmonary syndrome was coined to reflect the arterial hypoxemia which occurs in about one-third of patients with liver cirrhosis in the absence of detectable cardiorespiratory disease (Fig. 33.7).
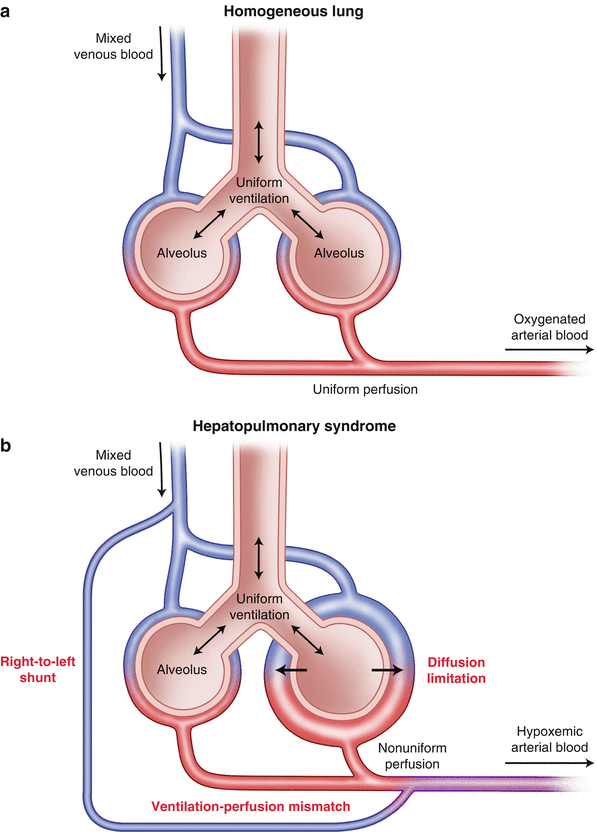
Fig. 33.7
(a) Normal pulmonary alveolar perfusion and diffusion; (b) three mechanisms of arterial hypoxemia in hepatopulmonary syndrome: Right to left shunt; diffusion limitation; ventilation-perfusion mismatch
The hepatopulmonary syndrome (HPS) is defined as the triad of liver disease and/or portal hypertension together with an increased alveolar-arterial gradient on room air, together with intrapulmonary microvascular vasodilatation [34, 35]. The intrapulmonary vascular dilatations result in a positive contrast echocardiogram, with echogenic material formed from agitated saline when injected intravenously passing from the right side of the heart to the left side with a 4–6-beat delay (Fig. 33.8) [34]. If the contrast crosses over to the left side faster than this then a septal defect should be considered.
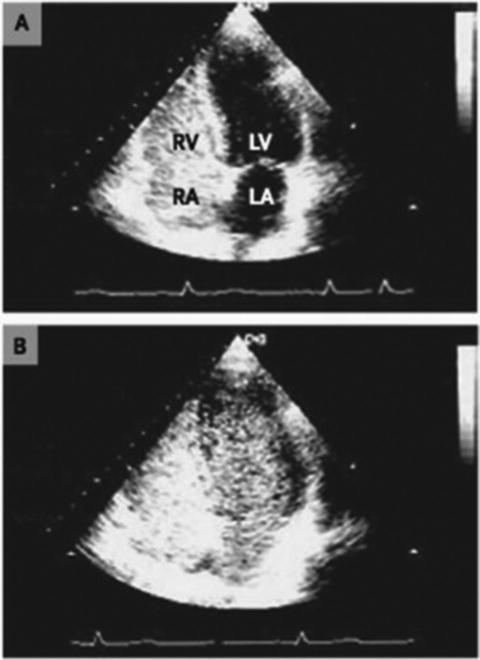
Fig. 33.8
Transthoracic echocardiogram demonstrating a delayed passage—5 to 6 beats—of echogenic material from the injection intravenously of agitated saline, from right to left heart
The impairment in oxygenation that occurs with HPS in some patients may be found to worsen upon standing. This is termed orthodeoxia and is a strong indicator of HPS [36, 37]. It is the result of preferential perfusion of the lung bases in a standing patient. Some patients with HPS also exhibit platypnea which is shortness of breath that is made worse by sitting up from the lying posture. This is the opposite of most other pulmonary conditions in which the patient breathes better on sitting up. Criteria defining HPS are outlined in Table 33.4 [34, 38]. Figure 33.9 shows albumin uptake in the lung, brain, and kidneys in a patient with hepatopulmonary syndrome [34].
Table 33.4
 Preoperative Evaluation and Preparation for Lung Transplantation
Anesthesia and Intraoperative Management of Renal Transplantation
Preoperative Evaluation and Preparation for Lung Transplantation
Anesthesia and Intraoperative Management of Renal Transplantation
 Intensive Care of the Deceased Multiorgan Donor: One Donor, Nine Lives
Intensive Care of the Deceased Multiorgan Donor: One Donor, Nine Lives
 Preoperative Liver Recipient Evaluation and Preparation
Preoperative Liver Recipient Evaluation and Preparation
 Technical Innovation and Visceral Transplantation
Postoperative Care of Heart Transplant Patients
Technical Innovation and Visceral Transplantation
Postoperative Care of Heart Transplant Patients




HPS criteria
Related posts:
Preoperative Evaluation and Preparation for Lung Transplantation
Anesthesia and Intraoperative Management of Renal Transplantation
Intensive Care of the Deceased Multiorgan Donor: One Donor, Nine Lives
Preoperative Liver Recipient Evaluation and Preparation
Technical Innovation and Visceral Transplantation
Postoperative Care of Heart Transplant Patients
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
