129 Pathophysiology of Sepsis and Multiple Organ Dysfunction
 Pathophysiology of Sepsis
Pathophysiology of Sepsis
Sepsis has been defined as an invasion of microorganisms or their toxins into the bloodstream, together with the host response to this invasion.1 Thus, the pathophysiology of sepsis combines the impact of infection with the host response of generalized inflammation, which finally leads to multiorgan dysfunction and death. This definition has been extended by the addition of several terms to more carefully describe the disease and its pathophysiology (Table 129-1). The American College of Chest Physicians/Society of Critical Care Medicine (ACCP/SCCM) Consensus Conference defined sepsis as a systemic inflammatory response syndrome (SIRS) caused by infection.2 More recently it has been recognized that SIRS is counteracted by a hypoinflammatory state that also plays an important role in the further development of organ dysfunction.3
| Term | Definition |
|---|---|
| Bacteremia | Presence of viable bacteria in the blood |
| Systemic inflammatory response syndrome (SIRS) | Generalized hyperinflammatory response to several impacts |
| Sepsis | SIRS caused by infection |
| Severe sepsis | Sepsis associated with organ dysfunction |
| Septic shock | Sepsis associated with arterial hypotension |
Data from ACCP/SCCM Consensus Conference Committee. Definition for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med 1992;20:864–74.
Sepsis is characterized by loss of hemostatic balance and endothelial dysfunction, which in turn severely compromise the cardiocirculatory system as well as intracellular homeostasis. Cellular hypoxia and apoptosis (programmed cell death) then contribute to organ dysfunction and death. The network of organ systems affected by sepsis is depicted in Figure 129-1.
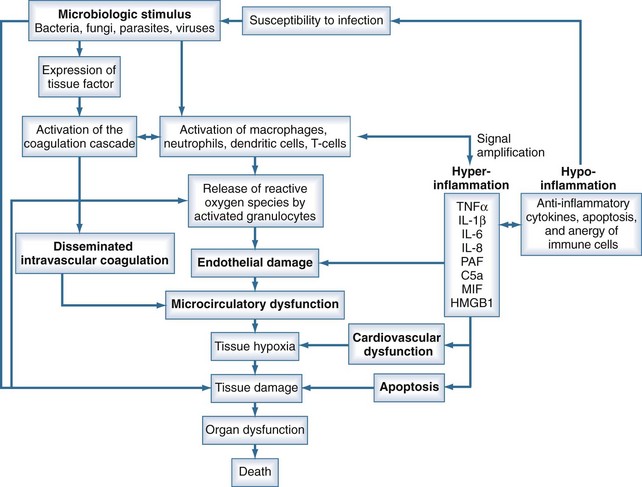
Microbiological Stimulus
By definition, infection is a fundamental part of the pathophysiology of sepsis. Any microorganism able to induce infection in humans may be complicated by sepsis. Bacteria as well as fungi, parasites, and to a lesser degree viruses can trigger the mechanisms that lead to sepsis. Although SIRS is the final common pathway of this process, the signal transduction pathway from infection to complex host response differs with the microbiological stimuli. Induction of an innate immune response is triggered by specific microbial molecules (e.g., bacterial wall components, exotoxins, bacterial DNA, viral RNA) called pathogen-associated molecular patterns (PAMPs). Damage-associated molecular patterns (DAMPs) are the noninfectious equivalents to PAMPs. DAMPs are released after cellular injury of the host (i.e., trauma) and can also induce the innate immune response.4
The presence of PAMPs is sensed by recognition molecules called pattern-recognition proteins (PRR), which are able to initiate a host response. These proteins may be categorized into secreted, transmembrane, and cytosolic PRRs. The Toll-like receptors (TLRs) represent the membrane PRRs. Eleven different TLRs have been discovered in mammals, whereas TLR-11 is not expressed in humans (Table 129-2). Retinoic acid-inducible gene I (RIG-I)–like receptors (RLRs) and the nucleotide-binding domain and leucine-rich repeat-containing receptors (NLRs) are cytosolic PRRs. RLRs recognize viral RNA and some double-stranded DNA. NLRs represent a large family of intracellular sensors that can detect pathogens and stress signals. NLRs detect microbiological products such as peptidoglycans and other degradation products of microorganisms as well as stress-related substances.5,6
TABLE 129-2 Human Toll-Like Receptors and Their Natural Ligands
| TLR Type | Related Pathogen-Associated Molecular Pattern | Location |
|---|---|---|
| TLR1 (via TLR2) | Bacterial products such as tri-acyl lipopeptides | Cell surface |
| TLR2 | Gram-positive bacterial products, including peptidoglycans; some virus-related proteins | Cell surface |
| TLR3 | Viral double-stranded RNA | Endosomal |
| TLR4 | Endotoxin, other bacterial products, some fungal products | Cell surface |
| TLR5 | Flagellin | Cell surface |
| TLR6 (via TLR2) | Some bacterial products | Cell surface |
| TLR7 | Single-stranded RNA | Endosomal |
| TLR8 | Viral single-stranded RNA | Endosomal |
| TLR9 | Viral and bacterial DNA | Endosomal |
| TLR10 | Unknown |
TLR, Toll-like receptor.
Gram-Negative Sepsis
In gram-negative bacteremia, initiation of the immune response is mediated primarily by lipopolysaccharide (LPS), a bacterial cell wall product. In plasma, LPS is bound to the LPS binding protein (LBP). Bound LPS is transported to the opsonic receptor, CD14, which is located on several cell membranes including on monocytes.7 A soluble form of CD14 interacts with CD14-negative cells (e.g., dendritic cells). However, CD14 alone cannot explain the actions of LPS, because CD14 does not have an intracellular tail.
Another binding site of LPS is the transmembranous receptor, TLR4, which exists in combination with the accessory protein, MD2.8 The binding of LPS to CD14 and TLR4 induces, via other molecules, activation of the transcription factor, nuclear factor kappa-B (NF-κB). Activated NF-κB migrates into the nucleus where it binds to and activates gene promoters, resulting in the transcription and expression of genes for cytokines and other proinflammatory mediators.9 In monocytes, LPS also induces cytokine transcription via the triggering receptor expressed on myeloid cells-1 and the myeloid DAP12-associated lectin.10 Intracellular pattern-recognition proteins in monocytes for LPS have recently been identified as another pathway of cytokine expression and include nucleotide-binding oligomerization domain 1 and 2 as LPS binding sites.11
Gram-Positive Sepsis
During the last decade, gram-positive bacteria have gained greater importance as causative organisms for sepsis.12 Gram-positive bacteria lack endotoxin and are recognized by cell wall components such as peptidoglycans and released bacterial toxins (exotoxins). Recently, lipoteichoic acid (LTA), a component of the cell wall in all gram-positive bacteria, has been recognized as the main pattern for recognition of gram-positive bacteria.13 TLR2 has been identified as the only pattern-recognition protein for gram-positive bacteria.14 The relationship between LTA and TLR2 is not completely clarified. Although LTA clearly interacts with TLR2, TLR2 is not a specific receptor for LTA, because TLR2 can recognize several other components of gram-positive bacteria.15 Gram-positive and gram-negative sepsis are indistinguishable clinically, suggesting a similar pathway of signal transduction. Indeed, peptidoglycans and LTA stimulate the release of tumor necrosis factor alpha (TNF-α), interleukin (IL)-6, and IL-10. It has been speculated that CD14 is also involved in the signaling of gram-positive infections.
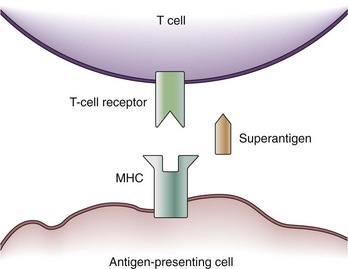
Some exotoxins cause a special type of septic shock called the toxic shock syndrome (TSS). TSS may be caused by the exotoxin, TSS toxin-1, staphylococcal enterotoxins from Staphylococcus aureus, or streptococcal pyogenic exotoxins.16 These toxins are capable of acting as so-called superantigens which deploy their effects via the T-cell antigen receptor (TCR). The TCR consists of five variable elements: Vβ, Dβ, Jβ, Vα, and Jα. Conventionally, the T cell is activated if the major histocompatibility complex (MHC) of an antigen-presenting cell matches all five elements. Thus, T cells are activated by proper antigen contact only. This results in the stimulation of about 1 in 10,000 T cells. However, a superantigen such as TSS toxin-1 works as a bridge between the MHC and the Vβ chain of the TCR only (Figure 129-2).17 Because T-cell activation now occurs independently of a match between the MHC and TCR, about 20% of the entire T-cell pool may be activated at once. Besides further T-cell proliferation, T-cell activation causes the release of several cytokines (i.e., interferon gamma [IFN-γ], IL-2, TNF-α) from T cells, as well as IL-1β and TNF-α from macrophages. Thus, the presence of superantigens results in a release of cytokines similar to gram-negative sepsis. It is assumed that actions other than cytokine production may be responsible for actions of superantigens in TSS; for example, superantigens may amplify the effects of LPS.
Other Microbiological Stimuli of Sepsis
Sepsis can also be induced by fungi, viruses, and parasites. Signal transduction by nonbacterial products, however, is not as well characterized as bacterial sepsis. In part, this may be due to the fact that induction of cytokine release differs markedly not only among different microorganisms but also among species. Nevertheless, the release of proinflammatory mediators has been demonstrated during infections with nonbacterial infections such as Candida albicans18 and Plasmodium falciparum.19 The signal transduction in viral infections is complicated by the fact that viruses can interfere with TNF-related cytokine release to avoid the host’s antiviral activities.20 Pattern recognition of viruses occurs mainly via endosomal TLR receptors which detect single- and double-stranded RNA or DNA (see Table 129-2).
The Immune Response in Sepsis
The cytokines TNF-α and IL-1β are released by activated macrophages and CD4 T cells within the first hour after infection. These primary mediators induce the release of several secondary mediators that amplify inflammation (Table 129-3). An important step in signal amplification is activation of the complement system. Besides being activated by antigen-antibody complexes, the complement system may be stimulated by bacterial surface sugars and endotoxin. The complement fragment C5a, a cleavage product of the complement cascade, is a strong chemoattractant. C5a appears about 2 hours after the initiation of sepsis and stimulates macrophages to further produce proinflammatory mediators. Another mediator that amplifies the immune response is macrophage migration inhibitory factor (MIF), which is produced by T cells, macrophages, monocytes, and pituitary cells in response to an infectious stimulus. MIF appears about 8 hours after the onset of sepsis and activates T cells and macrophages to produce proinflammatory mediators. About 24 hours after the initiation of sepsis, levels of high-mobility group box 1 (HMGB1) protein increase and appear to play a role in endotoxin-related sepsis. HMGB1 is a nuclear binding protein that, among other things, is capable of activating NF-κB. As a rather late mediator in sepsis, it is produced by macrophages and neutrophils and stimulates phagocytic cells.21
TABLE 129-3 Macrophage Mediators Involved in the Pathogenesis of Sepsis
| Mediator | Typical Effects |
|---|---|
| Cytokines: IL-1, IL-6, IL-12, IL-15, IL-18, TNF, MIF, HMGB1, IL-10 | Activate neutrophils, lymphocytes, and vascular endothelium; up-regulate cellular adhesion molecules; induce prostaglandins, nitric oxide synthase, and acute-phase proteins; induce fever |
| IL-10 is predominantly a negative regulator of these effects. | |
| Chemokines: IL-8, MIP-1α, MIP-1β, MCP-1, MCP-3 | Mobilize and activate inflammatory cells, especially neutrophils; activate macrophages |
| Lipid mediators: platelet-activating factor, prostaglandins, leukotrienes, thromboxane, tissue factor | Activate vascular endothelium; regulate vascular tone; activate extrinsic coagulation cascade |
| Oxygen radicals: superoxide and hydroxyl radicals, nitric oxide | Antimicrobial properties; regulation of vascular tone |
HMGB, high-mobility group B protein; IL, interleukin; MCP, monocyte chemoattractant protein; MIF, migration inhibitory factor; MIP, macrophage inflammatory protein; TNF, tumor necrosis factor.
Data from Cohen J. The immunopathogenesis of sepsis. Nature 2002;420:885–91.
It is not completely understood why inflammation becomes generalized in some patients but stays localized in others. Genetic variants of cytokines may play a role in this issue. Single nucleotide polymorphisms (SNPs) are single-base changes in the DNA which do not cause obvious changes in the function of the respective cytokine. However, SNPs in some cytokines are associated with a worse outcome from septic shock or an increased risk for developing sepsis.22–24 Among several others, such variants have been described in TNF-α, IL-6, and CD14.25 However, results from these studies are difficult to interpret because of contradictory results and differences in populations of different ethnicities.
The immune response in sepsis does not involve only proinflammatory mediators. As in many other physiologic processes, the organism produces inhibitors to control certain reactions. Proinflammatory mediators are counteracted by antiinflammatory molecules such as IL-4 and IL-10 because CD4 T cells can switch from the production of inflammatory cytokines (type 1 helper T cells [TH1]) to the production of antiinflammatory cytokines (type 2 helper T cells [TH2]). Soluble TNF receptors and IL-1 receptor antagonists (IL-1Ra) are released to inhibit the actions of TNF and IL-1 in their roles as primary mediators of sepsis. T cells, neutrophils, and macrophages also may become unresponsive to infectious stimuli (anergy).26 Another mechanism of the antiinflammatory response is the onset of apoptosis, a genetically programmed autodestructive release of proteases that induces cell death. In sepsis, enhanced apoptosis causes loss of immune effector cells, including CD4 and CD8 T cells, B cells, and dendritic cells.27 Absolute lymphocyte counts are significantly decreased in patients with sepsis.28 Further, apoptotic cells impair the function of surviving immune cells.29
Results from animal studies suggest that the autonomic nervous system is also involved in suppression of cytokine release during sepsis. In the experimental setting, vagal stimulation can inhibit TNF expression. It is hypothesized that an inflammatory reflex, with the afferent vagal nerve sensing cytokine release and an efferent immunosuppressing cholinergic arm, exists.30 The importance of such a reflex in humans merits further investigation.
The antiinflammatory response in sepsis has been termed the compensatory antiinflammatory response syndrome (CARS).3 It has been suggested that the first response to infection is hyperinflammation, which is followed by a hypoimmune state. From there, recovery would be possible, but the prolonged inability to eradicate microorganisms might result in the death of the patient.31 However, serum levels of antiinflammatory cytokines are increased in parallel with the increase of proinflammatory mediators.32,33 Thus, diminished inflammation develops at the same time as the process of hyperinflammation. Although the persistence of high levels of antiinflammatory mediators may contribute to mortality in septic patients, the clinical role of change between hyperinflammatory and hypoinflammatory states remains unclear.
Loss of Hemostatic Balance
Under normal conditions, the vascular luminal surface has anticoagulant properties. Tissue factor is a 4.5-kD protein that is bound to cell membranes which are normally not in contact with blood. Expression of tissue factor mainly depends on release of IL-6.34,35 Tissue factor expression occurs on mononuclear cells, but endothelial cells, polymorphonuclear cells, and other cell types may be additional sources. The expression of tissue factor induces intravascular thrombin formation initiated by the extrinsic coagulation pathway. Because this process is not restricted to a local area, it is called disseminated intravascular coagulation (DIC); DIC causes a consumption of coagulation factors.
Physiologically, excessive coagulation is counteracted by several natural anticoagulants including antithrombin, the thrombomodulin/protein C/protein S system, and tissue factor pathway inhibitor. In addition to the activation of tissue factor-dependent thrombin generation, anticoagulant function is attenuated in sepsis. Patients with sepsis demonstrate reduced levels of protein C and antithrombin due to consumption and reduced synthesis.36 Thus, the physiologic balance between procoagulant and anticoagulant substances is altered in sepsis as there is a shift of the hemostatic balance toward a procoagulant state (Figure 129-3).
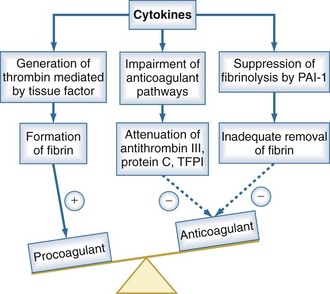
Besides its anticoagulant actions, the protein C pathway is an important link between coagulation and inflammation, because activated protein C has antiinflammatory properties. Protein S can bind to receptors that mediate an antiinflammatory regulatory loop of dendritic cell and monocyte inflammatory function. Thrombomodulin has been described to prevent excessive complement activation.37 Thus, there is considerable cross-talk between inflammation and coagulation which is impaired in sepsis as a result of depletion of the thrombomodulin/protein C/protein S system. There is also evidence that antithrombin blunts activation of several cytokines, suggesting that low antithrombin levels also affect the relationship between coagulation and inflammation.38
Endothelial Dysfunction
The endothelium produces several vasoactive mediators, including nitric oxide (NO), prostacyclins, and endothelin. NO is a potent vasodilator produced by NO synthase (NOS) from the amino acid, L-arginine. NO directly relaxes the vessel’s smooth muscle. There are two different forms of endothelial NOS: the constitutional form (cNOS) and the inducible form (iNOS). Physiologically, cNOS—also referred to as endothelial NOS (eNOS)—produces only small amounts of NO, and iNOS is expressed at low levels.39 In sepsis, iNOS expression is stimulated by cytokines such as IL-1β and TNF-α.40 This is followed by massive NO production and profound vasodilatation. Whether increased activity of cNOS also plays a role in sepsis is currently a matter of debate.
During inflammation, endothelial cells express adhesion molecules on their surface, which causes the adherence of leukocytes. These adhesion molecules include endothelial leukocyte adhesion molecule-1, intracellular adhesion molecule-1, and vascular cell adhesion molecule-1. Endothelial leukocyte adhesion molecule-1 is a selectin that mediates the initial step of leukocyte adhesion, followed by leukocyte rolling along the endothelial surface. The leukocyte finally migrates through the endothelial layer into the tissue, mediated by intracellular adhesion molecule-1 and vascular cell adhesion molecule-1 expression on both endothelial cells and leukocytes.41
Migration of leukocytes into the tissue is a physiologic mechanism to move immune cells to the site of infection. However, in generalized inflammation such as in sepsis, endothelial cells in several organs remote from the site of infection express adhesion molecules, inducing a generalized rolling and sticking of circulating leukocytes to the vascular surface. Adherence to endothelial cells activates leukocytes and induces a respiratory burst.42 The respiratory burst involves the release of cytotoxic substances such as elastase, myeloperoxidase, and reactive oxygen species. These products are capable of damaging endothelial cells and the surrounding tissue. Endothelial cell damage causes capillary leakage whereby intravascular fluid penetrates the extracellular space, leading to tissue edema.
Cardiac and Circulatory Dysfunction
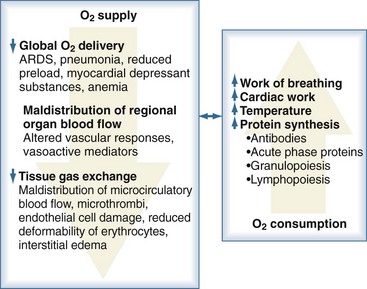
Sepsis is frequently complicated by organ dysfunction and shock. Shock occurs when the cardiovascular system is unable to transport sufficient amounts of oxygen to the tissues. In fact, sepsis compromises all levels of the cardiovascular system, resulting in cardiac dysfunction, vascular dysregulation, and microcirculatory damage. Impairment of the cardiovascular system causes a characteristic hemodynamic pattern that in cases of adequate fluid loading and the absence of severe preexisting cardiac dysfunction, consists of a high cardiac output, arterial hypotension, and low systemic oxygen (O2) extraction. In early sepsis, O2 consumption is increased owing to higher metabolic needs (i.e., tachypnea, fever, increased cardiac work, increased rate of protein synthesis), further compromising the relationship between O2 supply and demand (Figure 129-4). The hepatic and splanchnic region is markedly affected by these changes associated with sepsis. Hepatosplanchnic O2 uptake increases markedly during fever and bacteremia.43
< div class='tao-gold-member'>
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


