Fig. 1
Dimensions of pain
1 Pain-Related Amygdala Circuitry
The amygdala is closely interconnected with numerous cortical, subcortical, and brainstem areas. Figure 2 shows key amygdala nuclei and their connections relevant to sensory and pain-related processing. The lateral–basolateral nuclei (LA-BLA) form the input region for sensory, including nociceptive, information from thalamus (posterior areas) and cortical areas such as insula, anterior cingulate cortex, and other medial prefrontal cortical areas (Orsini and Maren 2012; Pape and Pare 2010; Marek et al. 2013; Price 2003). BLA projections of the medial prefrontal cortex (mPFC) provide emotion- and value-based information to guide executive functions such as decision-making and behavior control (McGaugh 2004; Holland and Gallagher 2004; Laviolette and Grace 2006). The BLA contains neurons that respond preferentially to noxious stimuli (Ji et al. 2010).
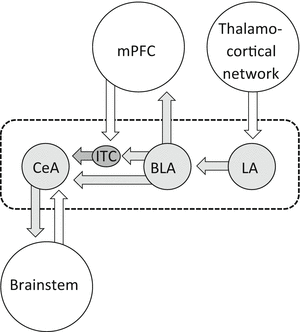
Fig. 2
Amygdala circuitry and interactions with cortical systems and brainstem. Input regions (lateral and basolateral amygdala, LA, BLA) process and transmit polymodal sensory and nociceptive information from thalamocortical systems to the amygdala output region (central nucleus, CeA) through direct excitatory projections or indirect feedforward inhibition involving interneurons in the intercalated cell mass (ITC). BLA forms close connections with mPFC that involve inhibitory interneurons in the cortex resulting in feedforward inhibition of mPFC principal cells. mPFC output neurons can engage ITC cells to control amygdala output. Dashed line indices area of the amygdala
Highly processed information generated in the LA-BLA network is transmitted to the central nucleus (CeA), which serves major amygdala output functions and projects to pain modulatory systems through forebrain and brainstem connections (Mason 2005; Neugebauer et al. 2004; Bourgeais et al. 2001; Price 2003) (Fig. 2). The laterocapsular division of the CeA (CeLC) receives nociceptive-specific information from the spinal cord and brainstem (external lateral parabrachial area) through the spino-parabrachio-amygdala pain pathway (Gauriau and Bernard 2002). The vast majority of CeLC neurons respond exclusively or predominantly to noxious stimuli and have large bilateral, mostly symmetrical receptive fields (Neugebauer et al. 2004, 2009). These CeLC neurons show non-accommodating spike firing properties characteristic of medium-size spine-laden peptidergic or GABAergic Type A projection neurons with targets in the brainstem, including PAG, and forebrain (Schiess et al. 1999; Jongen-Relo and Amaral 1998; Sun and Cassell 1993). Peptidergic (CRF or enkephalin containing) CeA projection neurons are innervated by calcitonin gene-related peptide (CGRP) containing terminals from the parabrachial area (Schwaber et al. 1988; Dobolyi et al. 2005; Harrigan et al. 1994), which is consistent with the peptidergic nature of the spino-parabrachio-amygdala pain pathway.
Interposed between LA-BLA and CeA is a cluster of inhibitory interneurons in the intercalated cell mass (ITC cells) which serve as a gate keeper to control amygdala output (Pape and Pare 2010; Likhtik et al. 2008; Marek et al. 2013; Jungling et al. 2008). ITC cells that inhibit CeA neurons are the target of excitatory projections from the infralimbic mPFC (McDonald 1998; Busti et al. 2011; Amir et al. 2011; Pinard et al. 2012) and are activated during behavioral extinction of negative emotional responses (Orsini and Maren 2012; Herry et al. 2010; Pape and Pare 2010; Sotres-Bayon and Quirk 2010). Pharmacological or mPFC-driven activation of ITC cells can also inhibit pain-related CeLC output and behaviors (Ren et al. 2013).
2 Pain-Related Amygdala Plasticity
Electrophysiological studies in anesthetized animals have consistently found increases in background and stimulus-evoked activity of individual CeLC neurons (Neugebauer and Li 2003; Li and Neugebauer 2004b, 2006; Ji and Neugebauer 2007, 2009; Ji et al. 2009) and BLA neurons (Ji et al. 2010) in a model of arthritic pain and in CeA neurons in a neuropathic pain model (Goncalves and Dickenson 2012) (Fig. 3). These “multireceptive” amygdala neurons are activated more strongly by noxious than innocuous stimuli and likely serve to integrate and evaluate sensory–affective information in the context of pain (Neugebauer et al. 2004). Biochemical and electrophysiological changes were observed only in the right amygdala in models of inflammatory pain (Ji and Neugebauer 2009; Carrasquillo and Gereau 2007, 2008). In neuropathic pain a transient activity increase was observed in the left amygdala but a persistent change in the right amygdala (Goncalves and Dickenson 2012). Mechanisms of pain-related lateralization remain to be determined.
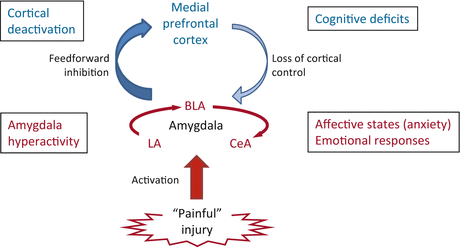
Fig. 3
Role of the amygdala in pain. Pain-producing events generate hyperactivity in the amygdala network of lateral, basolateral, and central nuclei (LA, BLA, CeA), which accounts for emotional–affective aspects of pain. Output from BLA deactivates medial prefrontal cortex through feedforward inhibition, resulting in cognitive deficits such as impaired decision-making. Decreased medial prefrontal cortical output to the amygdala allows the uncontrolled persistence of amygdala hyperactivity, hence persistence of pain
Neuronal activity changes in the amygdala are not simply a reflection of continued afferent input from spinal cord and other sources, but they result from an imbalance between excitatory and inhibitory synaptic mechanisms in the amygdala circuitry. Pain-related neuroplasticity in the amygdala has been established in electrophysiological and biochemical studies in brain slice preparations obtained from animals after the induction of different pain states, suggesting that brain changes persist at least in part independently of continued afferent input. Excitatory synaptic transmission to the CeLC is increased in acute models of arthritis (Neugebauer et al. 2003; Han et al. 2005; Bird et al. 2005; Fu and Neugebauer 2008; Fu et al. 2008), colitis (Han and Neugebauer 2004), and formalin-induced inflammation (Adedoyin et al. 2010) and in the spinal nerve ligation model of neuropathic pain (Ikeda et al. 2007; Nakao et al. 2012). Studies in the neuropathic pain model also reported right-hemispheric lateralization that depends on C-fiber-mediated inputs. Synaptic plasticity of parabrachial input to the CeLC is a consistent finding in various pain models, but there is also evidence for enhanced synaptic transmission at the LA-BLA (Ji et al. 2010) and BLA–CeLC synapses (Neugebauer et al. 2003; Fu and Neugebauer 2008; Ikeda et al. 2007; Ren and Neugebauer 2010; Ren et al. 2013).
Pain-related increased excitatory transmission and amygdala output can develop because inhibitory control mechanisms are impaired. Feedforward inhibition of CeLC neurons involves glutamatergic projections from BLA and mPFC to a cluster of GABAergic neurons in the intercalated cell masses (ITC cells). Decreased activation of this inhibitory gating mechanism in pain allows the development of glutamate- and neuropeptide-driven synaptic plasticity in the CeLC (Ren and Neugebauer 2010; Ren et al. 2013). A mechanism of impaired inhibition is the loss of cortical output (Ren et al. 2013) as the consequence of BLA hyperactivity that generates abnormally enhanced feedforward inhibition of principal cells in the mPFC (Ji et al. 2010; Ji and Neugebauer 2011). Failure to engage cortically driven ITC-mediated inhibitory control of amygdala processing may be a general mechanism of the abnormal persistence of emotional–affective states not just in pain (Apkarian et al. 2013; Ochsner and Gross 2005; Dalley et al. 2011).
Increased amygdala output as the result of neuroplasticity in the LA-BLA and CeLC has emerged as an important contributor to emotional–affective behaviors in animal pain models (Neugebauer et al. 2004, 2009). Decreasing amygdala activity with lesions or pharmacological interventions inhibits pain-related behaviors in different models (Han and Neugebauer 2005; Ren et al. 2013; Pedersen et al. 2007; Han et al. 2005; Manning 1998; Fu and Neugebauer 2008; Fu et al. 2008; Palazzo et al. 2008; Ji et al. 2010; Hebert et al. 1999). Importantly, increasing activity in the amygdala exogenously can exacerbate or generate pain responses under normal conditions in the absence of any tissue pathology (Han et al. 2010; Carrasquillo and Gereau 2007; Kolber et al. 2010; Qin et al. 2003; Myers et al. 2005; Myers and Greenwood-Van Meerveld 2010; Li et al. 2011; Ji et al. 2013). Increased amygdala activity is now also well documented in experimental and clinical pain conditions in humans (Liu et al. 2010; Baliki et al. 2008; Tillisch et al. 2010; Kulkarni et al. 2007; Simons et al. 2012).
3 Pharmacology of Pain-Related Processing in the Amygdala
The contributions of amino acid neurotransmitters (glutamate and GABA) and neuropeptides in the amygdala network relevant to pain-related processing are summarized in Fig. 4. A discussion of opioid function in the amygdala is beyond the scope of this article. There is strong evidence for a critical role of the amygdala, and the CeA in particular, in opioid-dependent pain modulation (Manning and Mayer 1995a, b; Zhang et al. 2013; Fields 2000; Manning 1998) and reward mechanisms (Bie et al. 2012; Cai et al. 2013), and μ-, κ-, and δ-opioid agonists can have direct effects on CeA amygdala neurons (Zhu and Pan 2004; Bie et al. 2009). However their role in pain-related amygdala processing and plasticity remains to be determined.
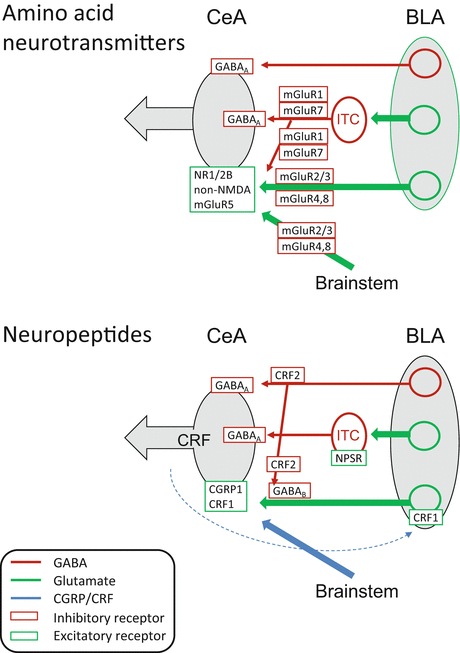
Fig. 4
Pharmacology of pain-related amygdala changes. Glutamatergic/GABAergic and peptidergic mechanisms are depicted in separate diagrams for the sake of clarity. BLA basolateral amygdala, CeA central nucleus of the amygdala, ITC intercalated cells, mGluR metabotropic glutamate receptor, NR1/2B NMDA receptor subunits NR1 and NR2B, NPSR neuropeptide S receptor. Dashed blue line indicates intra-amygdalar CRF release from CeA neurons
3.1 Ionotropic Glutamate Receptors
The CeLC receives glutamatergic inputs from the LA-BLA network and from brainstem areas (parabrachial input has been studied most extensively with regard to amygdala pain mechanisms). Activation of NMDA and non-NMDA receptors is required for the generation of hyperactivity of CeLC neurons in the arthritis pain model, because administration of antagonists for NMDA (AP5) or non-NMDA (NBQX) receptors into the CeA 6 h postinduction inhibited the increased background activity and evoked responses of these neurons in anesthetized animals (Li and Neugebauer 2004a). In the same experimental in vivo preparation, an NR2B-selective antagonist (Ro-256981) showed a different profile, inhibiting evoked but not background activity (Ji et al. 2009). In brain slices obtained from arthritic rats 6 h postinduction (acute stage), NMDA receptor-mediated transmission at the PB-CeLC synapse is increased through PKA-dependent phosphorylation of the NR1 subunit, and AP5 blocks the synaptic potentiation (Bird et al. 2005). In the subacute stage of the formalin pain model (24 h postinduction), NMDA receptors contribute to enhanced transmission at the PB-CeLC synapse (the NMDA receptor antagonist CPP inhibited transmission), but the NMDA/AMPA ratio is not increased compared to controls (Adedoyin et al. 2010). In contrast, synaptic plasticity of PB inputs to CeLC neurons in the spinal nerve ligation model of neuropathic pain (6–7 days postinduction) largely depends on non-NMDA but not NMDA receptors (Ikeda et al. 2007).
While the electrophysiological studies may suggest that NMDA receptors are more important for the induction rather than maintenance phase in models of inflammatory but not neuropathic pain, behavioral data argue against pain type- or stage-specific contributions. Bilateral intra-CeA injection of an NMDA receptor antagonist (MK-801) inhibited nocifensive (hind limb withdrawal reflex) and affective (place avoidance test) behaviors in the spared nerve injury model of neuropathic pain (Ansah et al. 2010). Even in normal animals, vocalization afterdischarges evoked by a noxious footshock (a measure of pain affect) were inhibited by antagonists for NMDA (AP5) or non-NMDA (CNQX) receptors administered into the CeA, whereas spinal reflexes (tail flick and hind limb movements) were unaffected (Spuz and Borszcz 2012). In this study bilateral administration of AP5 or CNQX was more effective than unilateral administration into either hemisphere.
3.2 Metabotropic Glutamate Receptors
The family of G-protein-coupled metabotropic glutamate receptors (mGluRs) comprises eight subtypes with splice variants, which can be classified into three groups (I–III) based on their sequence homology, signal transduction mechanism, and pharmacological profile.
Group I consists of mGluR1 and mGluR5 subtypes that couple to Gq/11 proteins to activate phospholipase C, resulting in the formation of IP3 to stimulate intracellular calcium release, generation of diacylglycerol (DAG) activating PKC, and activation of MAP kinases such as ERK (Nicoletti et al. 2011; Ferraguti et al. 2008; Niswender and Conn 2010). Electrophysiological, biochemical, and behavior data support an important role of mGluR1 and mGluR5 in pain-related amygdala neuroplasticity and amygdala-dependent behaviors. Antagonists for mGluR1 (CPCCOEt) and mGluR5 (MPEP) inhibited increased evoked responses of CeLC neurons recorded in anesthetized rats in a model of arthritis pain (Li and Neugebauer 2004b). MPEP, but not CPCCOEt, also inhibited enhanced background activity in the arthritis model and had inhibitory effects under normal conditions (Li and Neugebauer 2004b). Underlying mechanisms involve presynaptic mGluR1-mediated increase in excitatory and decrease in inhibitory transmission and postsynaptic mGluR5-mediated effects based on electrophysiological analyses in brain slices from arthritic rats (Ren and Neugebauer 2010; Neugebauer et al. 2003). In these studies selective mGluR1 antagonists (CPCCOEt and LY367385) inhibited excitatory but facilitated inhibitory transmission only in arthritis, whereas selective mGluR5 antagonists (MPEP and MTEP) inhibited synaptic transmission both under normal conditions and in arthritis.
Behavioral studies support an important role of amygdalar group I mGluRs in models of formalin-induced, arthritis, visceral, and neuropathic pain. Blockade of mGluR1 in the CeA with CPCCOEt inhibited stimulus-evoked audible and ultrasonic vocalizations and spinal reflexes that were increased in the kaolin/carrageenan arthritis pain model, whereas blockade of mGluR5 in the CeA with MPEP inhibited vocalizations but not spinal hind limb withdrawal reflexes. Vocalizations organized in limbic forebrain areas and particularly in the amygdala (measured as vocalization afterdischarges) were inhibited by both CPCCOEt and MPEP. Vocalizations during stimulation, which are organized at the brainstem level, were inhibited by CPCCOEt but not MPEP (Han and Neugebauer 2005). The results may suggest that mGluR1 and mGluR5 contribute to affective behaviors generated in the amygdala, whereas mGluR1, but not mGluR5, also regulate amygdala-dependent modulation of “sensory” nocifensive behaviors by descending brainstem systems. A similar pattern of differential contributions of mGluR1 and mGluR5 to amygdala functions was found in the spared nerve injury model of neuropathic pain. Application of antagonists for mGluR1 (CPCCOEt) or mGluR5 (MPEP) into the CeA inhibited aversive behaviors in the place avoidance test. CPCCOEt, but not MPEP, also inhibited hind limb withdrawal reflexes (Ansah et al. 2010). These studies were done in rats. Disruption of mGluR5 function in the CeLC of mice pharmacologically or with a conditional knockout approach inhibited paw withdrawal reflexes in the formalin pain model (Kolber et al. 2010) and visceromotor reflexes in the bladder distension pain model (Crock et al. 2012).
A possible conclusion emerging from these findings would be that mGluR1 contribute in a broader way to any type of pain behaviors generated or modulated by the amygdala, whereas mGluR5 function switches over time (in arthritis and neuropathic pain models) towards a preferential contribution to affective forebrain-mediated pain mechanisms. The electrophysiological data support this scenario because the presynaptic function of mGluR1 could affect the drive onto various amygdala output neurons, whereas the postsynaptic action of mGluR5 allows for the discrete activation of subsets of CeA neurons.
Under normal conditions, mGluR5 appears to mediate the facilitatory effects of group I mGluR activation in the CeA on vocalizations and spinal reflexes in the awake animal, on neuronal activity in the anesthetized preparation, and on synaptic transmission in amygdala brain slices (Neugebauer et al. 2003; Li et al. 2011; Ji and Neugebauer 2010; Li and Neugebauer 2004b). In these studies, the effects of an mGluR1/5 agonist (DHPG) were mimicked by a presumed mGluR5-selective agonist (CHPG) and/or blocked by a selective mGluR5 antagonist (MTEP). A selective mGluR1 antagonist (LY367385) inhibited the facilitatory effects of DHPG only on neuronal responses to visceral stimuli (colorectal distention) (Ji and Neugebauer 2010). The facilitatory effects of mGluR5 involved IP3-mediated calcium release to increase mitochondrial ROS production resulting in the activation of ERK1/2 and PKA, but not PKC. Inhibition of ERK (U0126) and PKA (KT5720) was necessary to block completely the excitatory effects of a ROS donor (tBOOH); a PKC inhibitor (GF109203X) had no effect (Li et al. 2011). ERK activation downstream of mGluR5 is also supported by studies in mice. DHPG administered into the CeA induced mechanical (decreased paw withdrawal thresholds) and visceral (response to bladder distension) hypersensitivity under normal conditions. The facilitatory effect was reduced by an mGluR5 antagonist (MPEP) or mGluR5 knockdown and was accompanied by ERK1/2 activation in the CeA (Kolber et al. 2010) and in the spinal cord (Crock et al. 2012).
Group II comprises mGluR2 and mGluR3 subtypes that couple negatively to adenylyl cyclase, cAMP, and PKA activation via Gi proteins (Nicoletti et al. 2011; Niswender and Conn 2010). Typically localized extrasynaptically they serve as presynaptic auto- or hetero-receptors (Niswender and Conn 2010). Pharmacological activation of group II mGluRs in the CeA with mGluR2/3 agonists (LCCG1 or LY354740) decreased the responses of CeLC neurons under normal conditions and in the kaolin-/carrageenan-induced knee joint arthritis pain model (Li and Neugebauer 2006). There was an increase in potency for inhibition of responses to noxious stimulation of the arthritic knee in the pain model but not for inhibition of responses to innocuous stimulation of the knee or of the intact ankle or background activity, suggesting perhaps an input- and activity-dependent effect, because noxious stimulation of the arthritic knee would likely generate the strongest input to the amygdala. A group II mGluR antagonist (EGLU) had no effect under normal conditions but increased the responses to stimulation of the knee in the arthritis pain state. This would be consistent with activity-dependent endogenous activation and gain of function of group II mGluRs in pain.
Electrophysiological analysis in brain slices showed that group II mGluRs act presynaptically to modulate synaptic plasticity in the amygdala in a model of arthritic pain (Han et al. 2006). A selective group II mGluR agonist (LY354740) inhibited excitatory transmission at the PB-CeLC synapse in slices from arthritic rats more potently than in controls without affecting neuronal excitability. A group II mGluR antagonist (EGLU) but not a GABAA receptor antagonist (bicuculline) reversed the inhibitory effect of LY354740. EGLU had no effect on its own, suggesting that the endogenous activation of group II mGluRs in the in vivo condition (see previous paragraph) may be due to inputs not preserved or active in the brain slice preparation. The pain-related function of group II mGluRs at the BLA–CeLC synapse remains to be determined, but group II mGluR agonists (LCCG1 and LY354740) inhibited excitatory transmission from BLA to CeLC under normal conditions, and the inhibitory effect persisted in the presence of antagonists for mGluR1 (CPCCOEt), mGluR5 (SIB-1893), GABAA (picrotoxin), and GABAB (CGP 55845) (Neugebauer et al. 2000).
The relative contribution of mGluR2 and 3 was examined in amygdala brain slices using an inhibitor (ZJ43) of the peptide neurotransmitter N-acetylaspartylglutamate (NAAG) that activates preferentially mGluR3 (Adedoyin et al. 2010). ZJ43 inhibited excitatory transmission at the PB-CeLC synapse more strongly than a group II mGluR agonist that does not distinguish mGluR2 and 3 (SLx-3095-1), suggesting a role for endogenous NAAG and perhaps a predominant contribution of mGluR3 under normal conditions. Both effects were blocked by a group II mGluR antagonist (LY341495). In the formalin pain model (24 h postinduction), ZJ43 was much less effective than SLx-3095-1 in reducing excitatory synaptic transmission, which would be consistent with a loss of function of NAAG or an increase in the contribution of mGluR2 relative to mGluR3. Electrophysiological effects correlated with behavior because systemic injection of ZJ43 blocked formalin pain-related synaptic plasticity in the CeLC as well as mechanical allodynia (Adedoyin et al. 2010).
Group III consists of mGluR4, 6, 7, and 8 subtypes. With the exception of mGluR6, which is found only in retinal ON-bipolar cells, they couple negatively to adenylyl cyclase, cAMP and PKA activation via Gi proteins like group II mGluRs (Nicoletti et al. 2011; Niswender and Conn 2010). Predominantly presynaptic receptors, mGluR4 and mGluR8 have high affinity for glutamate and are localized extrasynaptically on glutamatergic terminals, whereas low affinity mGluR7 is found in or near the active zone of the synapse on GABAergic terminals (Niswender and Conn 2010).
Evidence from electrophysiological studies using the prototypical broad spectrum agonist (LAP4) suggests that group III mGluRs can inhibit pain-related amygdala neuroplasticity. LAP4 inhibited evoked responses of CeLC neurons recorded in anesthetized rats more potently in the arthritis pain state than under normal conditions, whereas the inhibitory effect on background activity did not change (Li and Neugebauer 2006). A group III mGluR antagonist (UBP1112) had no effect under normal conditions but facilitated the evoked responses in arthritic rats. Electrophysiological analysis in brain slices showed that LAP4 decreased excitatory transmission at the PB-CeLC synapse more potently in the arthritis pain model than in control slices, and the inhibitory effects involved a presynaptic site of action (Han et al. 2004). UBP1112 reversed the inhibitory effect of LAP4 but had no effect on its own, which is different from the in vivo situation where the facilitatory effects of the antagonist suggest the possibility of endogenous receptor activation in the pain model. A likely explanation would be the requirement of amygdala inputs not present or active in the slice preparation. A group III agonist (LAP4) also inhibited transmission at the BLA–CeLC synapse under normal conditions, but pain-related function of group III mGluRs at this synapse remains to be determined.
The recent availability of subtype selective agonists allowed a more detailed analysis that showed opposing functions of mGluR7 and mGluR8 in the amygdala related to pain processing and modulation (Palazzo et al. 2008; Ren et al. 2011). Activation of mGluR7 with AMN082 increased CeLC output (synaptically evoked spiking) in brain slices from control rats but not from arthritic rats (Ren et al. 2011). AMN082 acted presynaptically to inhibit glutamate-driven synaptic inhibition of CeLC neurons (feedforward inhibition from BLA), but not monosynaptic inhibitory transmission, implicating BLA-activated GABAergic interneurons such as intercalated cells (ITC; see Sect. 4.2). In support of this interpretation, the effect of AMN082 was occluded in the presence of bicuculline to block GABAA receptors. The mGluR7-mediated disinhibition facilitated the excitatory drive of CeLC neurons. Thus, mGluR7 would act as a gatekeeper to regulate information flow to the CeLC by releasing GABAergic control, hence permitting excitatory inputs from the LA-BLA network to reach the CeLC under normal conditions. In contrast, a selective mGluR8 agonist (DCPG) inhibited excitatory transmission at the BLA–CeLC synapse and output of CeLC neurons more potently in brain slices from arthritic rats than under normal conditions. The mechanism was presynaptic on glutamatergic terminals and did not involve GABAergic modulation; DCPG had no effect on inhibitory transmission (Ren et al. 2011). The direct inhibitory effect of mGluR8 and the disinhibition by mGluR7 could be blocked with a group III antagonist (MAP4).
Behavioral data are consistent with these electrophysiological findings (Palazzo et al. 2008). Activation of mGluR7 in the CeA of normal rats with AMN082 facilitated spinal reflexes (withdrawal thresholds) and supraspinally organized affective responses (vocalizations); AMN082 also increased anxiety-like behavior (decreased open-arm preference in the elevated plus maze). The facilitatory effects of AMN082 were not detected in arthritic rats showing increased pain behaviors. Intra-CeA administration of an mGluR8 agonist (DCPG) had no effect in normal animals but inhibited the increased spinal reflexes and vocalizations of arthritic rats and had anxiolytic-like effects in these animals tested in the elevated plus maze (Palazzo et al. 2008). DCPG also decreased thermal hypersensitivity in a carrageenan-induced hindpaw inflammation model and the antinociceptive effect was blocked with a group III mGluR antagonist (MSOP) (Palazzo et al. 2011). Furthermore, an increase in mGluR8 gene, protein, and staining, the latter being associated with vesicular GABA transporter-positive profiles, has been found in the CeA after carrageenan-induced inflammatory pain. These results show that stimulation of mGluR8, which was overexpressed within the CeA in inflammatory pain conditions, inhibits nociceptive behavior. Such an effect is associated with an increase in 5-HT and Glu release, a decrease in GABA, and the inhibition of ON- and the stimulation of OFF-cell activities within RVM.
The results suggest that under normal conditions mGluR7, but not mGluR8, facilitates pain responses and has anxiogenic-like properties. In pain models, however, mGluR8, but not mGluR7, has inhibitory behavioral effects (Palazzo et al. 2008, 2011), and this functional change may involve upregulation of mGluR8 gene and protein expression (Palazzo et al. 2011).
3.3 GABA
The amygdala is rich in GABAergic neurons and GABA receptors, allowing the control of amygdala output through direct inhibition, feedforward inhibition, and disinhibition. Recent work has focused on a cluster of inhibitory interneurons in the intercalated cell mass (ITC cells) positioned between BLA and CeLC (Ren et al. 2013).
Evidence from electrophysiological studies in brain slices suggests that GABAergic transmission can control excitatory inputs and synaptically evoked outputs of CeLC neurons (Fu and Neugebauer 2008; Ren et al. 2011, 2013; Ren and Neugebauer 2010). However, GABAA receptor-mediated synaptic inhibition is lost or impaired in a model of arthritic pain. Decreased monosynaptic (Ren et al. 2011) and glutamate-driven disynaptic inhibition (feedforward inhibition sensitive to NBQX) (Ren and Neugebauer 2010; Ren et al. 2013) of CeLC neurons was found in brain slices from arthritis rats compared to controls. Feedforward inhibition can be evoked by stimulation of BLA output (Ren and Neugebauer 2010) or of medial prefrontal cortical fibers in the external capsule (Ren et al. 2013). ITC cells have been implicated in feedforward inhibition of CeLC neurons because pharmacological activation of ITC cells with neuropeptide S (NPS) (see Sect. 5) or high-frequency stimulation of excitatory external capsule input to ITC cells inhibited synaptic activation of CeLC neurons (Ren et al. 2013). These inhibitory inputs exert a tonic GABAergic tone on CeLC neurons under normal conditions but not in the arthritis pain model. Blockade of GABAA receptors with bicuculline in the CeLC under normal conditions facilitated excitatory transmission at the BLA–CeLC synapse through an indirect action in the network (Ren and Neugebauer 2010) and also increased synaptically evoked action potential firing, a measure of neuronal output (Ren et al. 2011). In brain slices from arthritic rats, however, bicuculline had no significant effect on excitatory synaptic responses of CeLC neurons, suggesting a loss of GABAergic inhibitory control that may contribute to the pain-related increase of excitatory transmission (Ren and Neugebauer 2010).
Behavioral data somewhat agree with the electrophysiological findings. In a model of neuropathic pain chronic constriction injury, bicuculline administered into the CeA had no effect on mechanical allodynia and hyperalgesia, but attenuated, rather than facilitated, affective pain behaviors (escape/avoidance test). The data would be consistent with a loss, and possibly even reversal, of tonic GABAergic inhibitory control (Pedersen et al. 2007). Exogenous activation of GABAA receptors in the CeA with muscimol, however, had antinociceptive effects and attenuated escape/avoidance behaviors, which would indicate the presence of functional GABAA receptors. Neurochemical studies found no evidence for significant changes of extracellular GABA levels in the BLA in the formalin pain model (Rea et al. 2009) and in the CeA in a model of carrageenan-induced hindpaw inflammation (Palazzo et al. 2011). In these studies, baseline rather than evoked release was studied. Electrophysiological data implicated mGluR1 in the reduced or lost GABAergic inhibition (see Sect. 4.1 Group I). The mechanisms of impaired GABAergic control of amygdala function remain to be determined.
3.4 Neuropeptide CGRP
CGRP is a 37-amino-acid peptide that binds to G-protein-coupled receptors, including CGRP1, which couple positively to adenylyl cyclase, cyclic AMP formation, and protein kinase A (PKA) activation (Wimalawansa 1996; Poyner et al. 2002; Van Rossum et al. 1997). Functional CGRP1 receptors are formed by a heterodimeric complex of the calcitonin receptor-like receptor (CRLR) and receptor activity-modifying protein 1 (RAMP1) (Robinson et al. 2009; McLatchie et al. 1998). Particularly high levels of CGRP (de Lacalle and Saper 2000; Schwaber et al. 1988; Kruger et al. 1988; Dobolyi et al. 2005), CGRP binding sites (Wimalawansa 1996; Van Rossum et al. 1997), and proteins (CRLR and RAMP1) required for functional CGRP1 receptors (Ma et al. 2003; Oliver et al. 2001) have been described in the amygdala (CeA). CGRP can interact with other receptors (Robinson et al. 2009; Hay 2007; Poyner et al. 2002) such as a putative CGRP2 receptor that may include RAMP2 or RAMP3 rather than RAMP1 (Hay 2007). Whereas RAMP1 dominates in the CeA, the BLA contains relatively more RAMP2 than RAMP1 (Oliver et al. 2001). The exclusive source of CGRP in the amygdala is the lateral parabrachial area, and CGRP-immunoreactive parabrachial fibers essentially delineate the CeLC (Schwaber et al. 1988; de Lacalle and Saper 2000; Kruger et al. 1988; Dobolyi et al. 2005), making CGRP a marker of parabrachial inputs to the “nociceptive amygdala.”
Electrophysiological and behavioral data show an important role of CGRP and CGRP1 receptors in the CeA in pain-related neuroplasticity and behaviors. Administration of selective CGRP1 receptor antagonists (CGRP8-37 and BIBN4096BS) into the CeA inhibited increased responses of CeLC neurons to mechanical stimulation of the arthritic knee and non-injured ankle in anesthetized rats with a kaolin-/carrageenan-induced knee joint arthritis (Han et al. 2005). The antagonists were more efficacious in the pain model than under normal conditions. In amygdala brain slices from arthritic rats, CGRP1 receptor antagonists inhibited synaptic plasticity of parabrachial inputs to the CeLC but had no significant effect under normal conditions (Han et al. 2005). Detailed electrophysiological analyses showed that CGRP1 receptors contribute to pain-related plasticity through a protein kinase A (PKA)-dependent postsynaptic mechanism that involves NMDA, but not AMPA, receptors. Pharmacological blockade of CGRP1 receptors in the CeA with CGRP8-37 inhibited spinal hind limb withdrawal reflexes and supraspinally organized pain behaviors (audible and ultrasonic vocalizations) of awake arthritic rats (Han et al. 2005). CGRP8-37 had no effect on these behaviors in normal animals without arthritis.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


