Chapter 6
Neurophysiology of pain
By the end of this chapter readers should have an understanding of:
1 The neurophysiology underlying somatic and visceral pain.
2 Mechanisms of sensitization in the peripheral and central nervous system.
3 Ways in which the brain controls nociceptive processing.
OVERVIEW
Interpretation of the pathophysiological aspects of a patient’s symptoms requires that the clinician understands the sensory nervous system’s function and plasticity. He also needs to be aware that pain, whatever its origin, tends to be interpreted by the patient as somatic and nociceptive. In other words, pain is usually subjectively felt in skin and musculoskeletal structures, even though it may be of, for instance, visceral or neurogenic origin. Additionally, the perception of pain and concomitant suffering is subject to changes in sensory processing. A grounding in pain physiology enables the clinician to consider whether a patient’s pain is indeed likely to be somatic in nature, or whether other factors may be causing, maintaining or altering it. Leading pain psychologist Chris Main states that ‘Disregard of the role of central pain-processing mechanisms leads to failure to integrate the contribution of cognitive factors, pain memories and the emotional impact of pain into the explanation given to the patient of their pain experience’ (Main 2009).
Finally, it is worth noting that most of the information in this chapter is based on laboratory findings. It is often plausible that these findings apply to the clinical situation because they closely resemble what we observe in and hear from patients. Clinicians must always be aware that human beings are extremely complex, however, and that laboratory research gives us models but no absolute certainty. Findings from experiments on a small part (e.g. dorsal horn cells in a rat) may or may not be reflected in the complex experience of live human beings. There is increasing call for translational pain research in order to bridge the gap between scientific research and clinical practice (Mao 2009).
NOCICEPTION OF SOMATIC AND VISCERAL ORIGIN
Cutaneous nociception
Generally, changes in the tissues such as pressure, heat or damage activate receptors at the peripheral terminal of sensory neurons. This activation produces one or more action potentials in the neurons, which are conducted to the central nervous system and eventually the brain. The brain receives precise information about tissue events because of the way stimuli are encoded and transmitted (Gardner et al 2000)*. First, most receptors are highly specialized for a specific type of stimulus. Second, the more intense the stimulus, the higher the frequency of the action potentials is. Finally, the way pathways in the central nervous system are organized reflects the origin of the stimuli (somatotopical organization).
Stimuli that are associated with actual or impending tissue damage are classed as noxious (see also Box 6.1). They activate sensory receptors called nociceptors, which have a high stimulation threshold and therefore only respond to stimuli outside the normal and healthy range. Nociceptors may be specialized for either extreme thermal or mechanical stimuli, or they may be polymodal (sensitive to extreme thermal and mechanical stimuli as well as chemicals. Once nociceptors are activated, they generate action potentials in unmyelinated C fibres and thin myelinated Aδ fibres.
Thermal and mechanical nociceptors form the terminals of Aδ neuronal fibres, while C fibres end in polymodal nociceptors (Basbaum & Jessell 2000). These fibres have different response characteristics and are associated with different aspects of pain (Box 6.2). Neurons of the Aδ type respond quickly and also have a short adaptation time, so they tend to be active only for the duration of the stimulation. They therefore produce a brief spike of activity. The pain associated with Aδ stimulation is sharp and brief. The receptive field is small so sensations are well localized. The conduction time of Aδ fibres is much faster than for C fibres, so their activity is perceived first (so-called first pain).
On the other hand, brief stimulation of C fibres leads to a slow development of action potentials after Aδ activity has ceased. Low conduction speeds lead to a second pain, while the large receptive field means that this pain is diffuse, i.e. difficult to localize with precision. Acute localized trauma may therefore be felt as a sharp localized initial pain, followed by a wave of deep dull aching pain (van Cranenburgh 2000, pp 85–88).
Apart from enabling sensation, nociceptive neurons play an active role in the regulation of inflammation. Their peripheral terminals store neuropeptides that have been produced in the cell body. These peptides, called substance P (SP) and calcitonin gene-related peptide (CGRP), may be released when nociceptive fibres are activated (Basbaum & Jessell 2000). This produces vasodilation and therefore an area of redness and oedema (flare). This ‘fuelling’ of local inflammatory processes does not take place if the peripheral nerve is blocked proximally (LaMotte et al 1991) and is known as neurogenic inflammation. The neuropeptides are also responsible for peripheral sensitization of nociceptors in the area by stimulating local release of histamine from mast cells. This creates allodynia and hyperalgesia to mechanical stimuli and heat in the area of flare (Raja et al 1984). The mechanical sensations affected are stroking and sharpness, but not blunt pressure (Meyer et al 2006).
Deep somatic nociception
Joint structures and muscles are innervated by Aβ, Aδ and C fibres (for an extensive review see Schaible 2009). Aβ fibres have specialized mechanoreceptors responding to changes in position, movement and activity (Gardner et al 2000). On the other hand, the nociceptive Aδ and C fibres have free nerve endings, which will normally respond only to extreme mechanical stimuli such as end of range overpressures (Schaible 2006). They will also signal tissue damage and inflammation because of their sensitivity to inflammatory mediators such as bradykinin and prostaglandins. Some C fibres form an exception in that they have a low mechanical stimulation threshold. They terminate in so-called ergoceptors, which play a role in homeostatic regulation in exercise (Mense 2008). Joint cartilage is not innervated.
Inflammation produces peripheral sensitization of nociceptors in the joint, producing mechanical allodynia (pain as a result of normally innocuous joint movements or stretch). Muscle nociceptors will respond to inflammation in a similar way, but also to muscle activity under ischaemic conditions. The latter applies to constriction induced in laboratory conditions, but probably also in exercise involving sustained or repetitive contractions. Mechanisms of central sensitization and descending control are probably similar to those described below, although the reader should be aware that research specifically addressing these processes is limited (Schaible 2006). There is evidence that repeated noxious unilateral muscle stimulation leads to central sensitization, which can last for several weeks after stimulation has ceased.
Visceral nociception
The viscera are very different from somatic structures in their innervation and pain perception, and there are differences between organs (refer to Mayer & Gebhart 1994 and Ness & Gebhart 1990 for reviews). Viscera have two sets of afferents: one set follows sympathetic nerves and parasympathetic sacral nerves back to the spinal cord, while the other follows the parasympathetic vagus nerve to the brain stem (Bielefeld & Gebhart 2006). It is important to note that the sympathetic and parasympathetic systems themselves are purely efferent, although they may be influenced by visceral afferents in the paravertebral ganglia or spinal cord.
Viscera are mostly innervated by Aδ and C fibres, which unlike cutaneous afferents respond to mechanical stimuli as well as the usual chemical and thermal sensations (Gebhart et al 2004). The types of stimuli which provoke pain are quite different from those causing somatic pain. For example, distention of the bowel and other hollow organs can be painful, while cutting the bowel does not cause any pain whatsoever (Cervero & Laird 1999).
Visceral activity is generally not perceived and nociceptive stimulation leads to diffuse pain that is difficult to localize (Mayer & Raybould 1990). The pain refers to more superficial structures (see below) and can therefore be mistaken as being somatic in nature. This is an important point for clinicians to bear in mind when questioning and examining a patient; internal pathologies may masquerade as musculoskeletal ones. In functional pain disorders such as non-cardiac chest pain and irritable bowel syndrome, the responses to normal visceral stimuli become enhanced, leading to hyperalgesia and referred pain (see, for example, Mertz et al 1995 and Sarkar et al 2000). These changes are the result of changes in sensory processing in the peripheral and central nervous system, rather than in the organs themselves.
Referred pain
Referred pain is pain felt at a distance from and in tissues distinct from the origin of nociceptive stimulation. It is a phenomenon associated with a non-pathological intact nervous system and must therefore be distinguished from neurogenic pains such as nerve root pain. The exact mechanisms are as yet uncertain, but it is thought that changes in sensory processing in the dorsal horn are responsible (Mense 2008). Referred pain is not purely subjective and may be associated with measurable hyperalgesia in several studies (Coutinho et al 2000; Sarkar et al 2000).
Pain referral can be said to be from deep to superficial structures (Arendt-Nielsen et al 2000): visceral pain tends to be felt in musculoskeletal and cutaneous tissues, while muscle pain is referred to the skin. Cutaneous pain, on the other hand, does not refer to other tissues.
Two theories offer the most plausible explanation for referred pain (see Fig. 6.1; Arendt-Nielsen et al 2000; McMahon 1997). The convergence–projection theory suggests that afferents from deep tissues share second-order neurons with more superficial tissues. The brain therefore has no way of establishing the exact origin of the pain. It interprets the pain as coming from the more superficial structures, presumably because this is where most sensation normally comes from. The convergence–facilitation theory poses that nociceptive input from deeper tissues may sensitize secondary neurons. Sensory input from superficial structures arriving at the same secondary neurons will be facilitated, so that innocuous sensations from those structures may now be perceived as painful.
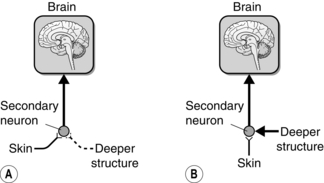
Fig. 6.1 The two main theoretical models of referred pain. (A) The convergence-projection model suggests that areas of skin and deeper tissues such as joints or viscera have an equal influence on the secondary neuron in the dorsal horn. (B) The convergence–facilitation model suggests that nociceptive stimulation from deeper structures creates pain by facilitating cutaneous input into secondary neurons in the dorsal horn.
There are two additional theories, each with their limitations. The thalamic convergence model suggests that sensory input from skin and viscera combines in the thalamus rather than the spinal cord. This model can account for several characteristics of referred pain, except its reduction following the application of a tourniquet or local anaesthetic (Laursen et al 1999). Projection of visceral afferents to the brain stem may therefore be partly responsible for referred pain. Finally, the axon reflex model is based on the assumption that some afferents are split to innervate both cutaneous and visceral structures. This type of axon is rare and therefore unlikely to be involved in the majority of cases of referred pain (McMahon 1997).
PERIPHERAL SENSITIZATION
It seems so intuitive that damaged or inflamed tissues will cause pain that few clinicians stop to wonder by what mechanism these processes stimulate sensory neurons. However, only if this is understood can a rational analysis be made of the possible origins of a patient’s pain. Apart from damage to peripheral nerves, the subject of Chapter 10, damaged cells release chemicals such as histamine, bradykinin, serotonin and prostaglandin, some of which are inflammatory mediators released as part of the inflammatory response. These chemicals can either stimulate nociceptors directly (i.e. make them depolarize) or sensitize them (i.e. bring the membrane potential closer to depolarisation threshold) so that other stimuli are more likely to activate them. The latter is known as peripheral sensitization. An example is a fresh bruise, which may not be felt spontaneously but which is acutely sensitive to local pressure (mechanical allodynia). It will also be more sensitive to painful stimuli (primary hyperalgesia, as opposed to secondary hyperalgesia, which is the result of changes in the dorsal horn).
An overview of the mechanisms involved in peripheral sensitization follows (for a comprehensive review see McMahon et al 2006). The peripheral terminal of a nociceptive neuron has three types of receptors. Ligand-gated ion channels respond to hydrogen ions (present in acidic conditions), ATP, heat or capsaicin (an extract of chilli peppers) by opening, creating an ion flux which depolarizes the cell thus creating action potentials. Prostaglandins, bradykinin, epinephrine (adrenaline) and ATP bind to G-protein receptors, which do not make the neuron fire but create internal chemical reactions. This leads to the sensitization of the ion channels as well as the sodium channels along the axon, thereby lowering the stimulation threshold and increasing the responsiveness of the neuron. Ligand-gated ion channels open in response to hydrogen ions (an extract of chilli peppers), creating an ion flux, which depolarizes.
The third type of receptor is tyrosine kinases, which respond to neurotrophic factors and cytokines. Small amounts of neurotrophic factors such as nerve growth factor (NGF) are released by tissues under normal conditions. They are important for the maintenance of the neurons and their make-up (see Chapter 10). Research suggests that NGF levels are increased during inflammation, sensitizing the nociceptive neurons and creating allodynia and hyperalgesia. The release of cytokines such as tumour necrosis factor-alpha (TNF-α) and interleukins (IL) also contributes to inflammatory pain. Their importance is confirmed by the efficacy of drugs that inhibit the action of TNF-α in the treatment of autoimmune diseases such as rheumatoid arthritis, ankylosing spondylitis and Crohn’s disease. As they do not cross the blood–brain barrier, their action must be peripheral (Marchand et al 2005).
CENTRAL SENSITIZATION
Interestingly, in the injury model described above the area demonstrating mechanical hyperalgesia is considerably larger than the area of flare (Box 6.4). This is not the result of a spreading of sensitization within the tissues because it develops even if a tight band restricts fluid movement (LaMotte et al 1991






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


