Chapter 46 Neonatal Respiratory Disease
Effective gas exchange within the lung requires both adequate ventilation and perfusion. Determinants of ventilation include the ventilatory “pump” (e.g., central drive, muscle strength, and chest wall recoil),1 compliance of the lung and chest wall, and resistance to airflow within the airways. Determinants of perfusion include the circulatory pump (right ventricular output) and pulmonary vascular resistance. A disorder in any one or a combination of these determinants can lead to respiratory insufficiency. Clearly then, a variety of disorders, either pulmonary or nonpulmonary, can lead to respiratory insufficiency.
Acute or Early-Onset Respiratory Disorders
Delayed Clearance of Fetal Lung Liquid
Fetal lung fluid, which is actively produced by the lung and is critical for normal fetal lung development,2 must be readily cleared from the airways to allow normal breathing after birth. On the basis of results from animal studies, the relative volume of liquid within potential airspaces, which remains constant in utero, is approximately 20 to 30 mL/kg near term and is the result of a balance between net accumulation within the lung (production minus reabsorption) and efflux out the trachea into the amniotic cavity.3 In the hours to days before delivery, net accumulation diminishes, and during labor, reabsorption predominates.4 As a result, extravascular lung liquid (i.e., liquid within the airspaces and interstitium) decreases. Any excess fluid remaining within the airspaces at the time of delivery is further removed as air entry into the lung displaces liquid from the airways into the interstitium. Residual liquid within the interstitium is then taken up into the circulation during the next several hours. Excessive extravascular liquid will lead to impairment of gas exchange, interstitial liquid pressure can compress small airways leading to atelectasis and gas trapping, and excess liquid within airspaces will impair alveolar gas exchange.5
Fetal lung liquid production and reabsorption are the result of active ion transport6 and are presumably hormonally regulated (Figure 46-1). Chloride ions enter the lung epithelial cell across the basolateral membrane via a Na/K/2CL cotransporter (the transporter on which furosemide acts). The mechanism of transepithelial movement of lung fluid at the time of birth is passive movement of sodium through epithelial sodium channels (ENaC), which are closed during fetal life; adrenergic stimulation during parturition activates these channels.7 Although β-adrenergic agents such as terbutaline and epinephrine enhance Na ions and thus liquid reabsorption,7,8 β-adrenergic blockade does not inhibit the reabsorption of lung liquid during spontaneous labor and delivery in animal studies.9,10 Other hormones of parturition, such as vasopressin, may be important as well.11,12
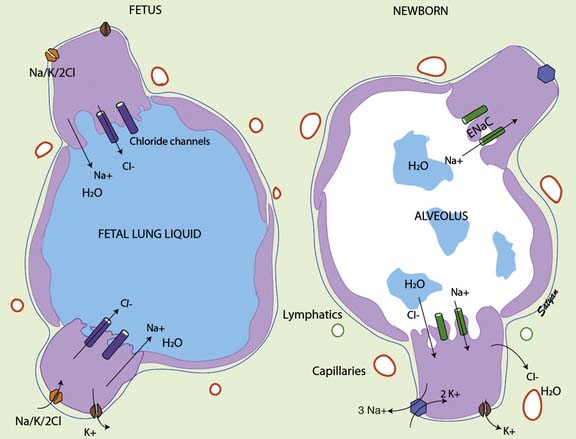
Figure 46–1 Mechanism of fetal and neonatal lung fluid transport.
(Modified from Guglani L, Lakshminrusimha S, Ryan RM: Transient tachypnea of the newborn, Pediatr Rev 29:e59-e65, 2008. Copyright Satyan Lakshminrusimha.)
The pulmonary circulation is also a key factor in fetal lung fluid clearance; not only does interstitial liquid drain directly into the circulation,13,14 but the dramatic increases in pulmonary blood flow seen after birth may enhance reabsorption of liquid from fetal airspaces.15 The onset of breathing not only increases the surface area for liquid reabsorption but also is associated with the opening of pores through which liquid can readily enter the interstitium.16 Drainage of interstitial liquid is generally complete by the end of neonatal transition (4 to 6 hours). Interstitial liquid appears to be directly absorbed into the microcirculation, and this process is governed by Starling forces; the contribution of lymphatic drainage is negligible.3
Interference with this process of liquid removal from the airspaces and interstitium leads to impaired gas exchange and respiratory distress with a variable clinical presentation. Excess liquid within the airspaces reduces compliance and increases intrapulmonary shunting, which results in tachypnea and mild to moderate hypoxemia. The chest radiograph may show opaque areas similar to neonatal pneumonia or surfactant-deficient respiratory distress syndrome (RDS). This picture is often described as “prolonged neonatal transition” or “delayed extrauterine adaptation.” Excess interstitial liquid reduces compliance and compresses small airways, which leads to the clinical signs of tachypnea and air trapping. The chest radiograph may show streaky densities within the lung, fluid collection within interlobar fissures, or even small pleural effusions; the lungs will also appear hyperinflated because of gas trapping. This clinical picture is often labeled retained fetal lung liquid or transient tachypnea of the newborn.5,17
Several additional points are worth noting. Infants who have been stressed in utero or whose mothers have been receiving β-mimetic agents are less likely to have retained lung liquid. Preterm infants with this condition may be mistakenly assumed to have surfactant-deficient RDS; supportive treatment is similar, although surfactant therapy has not been studied in infants with this condition. Because neonatal pneumonia also can present with tachypnea, an oxygen requirement, and a “wet” chest radiograph, infants with retained lung liquid who have symptoms often are treated with antibiotics. The clinical severity of this disorder can vary widely, and although most infants need only supplemental oxygen, others may require intubation and mechanical ventilation for 12 to 24 hours. Supplemental oxygen should be required for no more than 24 to 48 hours, but tachypnea may persist for several days.
It is of interest to note that infants born to mothers who have asthma are at increased risk of transient respiratory difficulty immediately after birth.18,19 In addition, reactive airway disease is more likely to develop later in life in newborns who present with transient respiratory distress.20,21
Pulmonary Air Leak Syndromes
Pulmonary air leak syndrome encompasses a spectrum of disease including pneumothorax, pneumomediastinum, pneumopericardium, subcutaneous emphysema, pneumoperitoneum, and pulmonary interstitial emphysema (PIE). As a group, pulmonary air leaks are more common during the neonatal period than at any other time of life. The two most common types of air leaks—pneumothorax and pneumomediastinum—occur spontaneously in 1% to 2% of term neonates.22 Preterm infants with surfactant-deficient respiratory distress had previously reported rates of air leaks in excess of 30%23; these rates fell rapidly with the advent of surfactant therapy in the 1980s but still remain around 5%.24 Infants with meconium aspiration or hypoplastic lungs have much higher rates of air leaks. Although techniques such as HFV and ECMO have markedly reduced the incidence of air leaks associated with these disorders, the rates still remain high, at 25% to 30%.25,26
The pathophysiologic cause of pulmonary air leaks has been hypothesized for many years as the uneven filling and redistribution of air in the lung; overdistension of more compliant air spaces leads to rupture. The alveoli most susceptible to this injury are those that border on arterioles and other structural elements of the lung; uniform protection by surrounding alveoli is lacking. After rupture of the alveolus or terminal airspace, air escapes into the lung interstitium and tracks along the vascular sheaths toward the hilum. For reasons that remain unclear, in the preterm infant the air may dissect within the interstitium (PIE).27 More commonly, the air breaks through the pleural reflections at the hilum (pneumomediastinum) and from there gains access to various potential compartments: the pleural cavity (pneumothorax), the pericardial reflection (pneumopericardium), and the soft tissue planes of the neck (subcutaneous emphysema), or across the diaphragmatic apertures and into the peritoneal space (pneumoperitoneum).
In the past, pulmonary air leaks most commonly occurred as a result of excessive ventilatory pressures due to either aggressive mechanical ventilation (barotrauma) or air trapping caused by partial airway obstruction with meconium or other debris (ball valving). Now, air leaks are more commonly seen during the recovery phase of acute respiratory disease, when lung compliance dramatically improves and pressure-limited ventilation leads to excessive tidal volumes (volutrauma). This phenomenon explains the clinical observations that air leaks tend to occur during the recovery phase of RDS28 and that the incidence of air leaks actually increased during early trials of surfactant therapy.29 Both observations underscore the need to closely monitor ventilatory volumes and wean pressure aggressively as compliance improves; these observations also suggest that volume-limited ventilation may be safer during the recovery phase of acute neonatal respiratory disease, although this theory has not been studied.
The approach to management of air leaks in infants is similar to that in older children. Pneumothorax, pneumopericardium, and occasionally pneumoperitoneum are acute, life-threatening emergencies because they can seriously impair cardiac output by decreasing venous return. A high degree of suspicion for air leaks in a patient who has a sudden cardiovascular deterioration for no apparent reason is critical for prompt diagnosis. Transillumination of the relatively translucent neonatal chest wall with an intensely focused light source is a quick and useful tool for diagnosing a large pneumothorax. Immediate aspiration of the air, preferentially with a large-bore angiocatheter, should be done without radiographic confirmation if the infant is severely compromised. Clinical signs of compromise, not the radiographic interpretation of percent size or extent, should dictate treatment, and many cases can be managed expectantly.30 An unstable or recurrent pneumothorax may require a thoracostomy tube; however, tube thoracostomy may have significant complications, including parenchymal lung injury, phrenic nerve paralysis, chylothorax, and hemorrhagic pericardial effusion.30 Penetration of the chest tube into the pulmonary parenchyma is a complication that often is not appreciated by chest radiographs; one study of autopsies of infants with pneumothorax reported a lung perforation rate of 25%.31 Cardiac tamponade resulting from a pneumopericardium may be suggested by distant heart tones and hypotensive shock with a normal-appearing electrocardiogram tracing (so-called electromechanical dissociation). Pneumomediastinum often is asymptomatic and rarely benefits from drainage, even in the presence of symptoms. PIE occurs predominantly in preterm infants and often leads to a vicious cycle of increasing ventilator delivery pressures to open alveoli compressed by extrinsic air, which in turn leads to more extravasation of air and further collapse. Conventional treatment of PIE, including positioning, selective mainstem intubation, and steroids, has been unsatisfactory.27,32 HFV, which may maintain alveolar patency while reducing inspiratory airway pressure, appears to be the ventilatory mode of choice in managing infants with PIE33 and refractory pneumothoraces.34
Pulmonary Hemorrhage
The term pulmonary hemorrhage is often misapplied in the neonatal intensive care setting. True pulmonary hemorrhage in a neonate is rare and almost alwaysresults in death. In most cases, what is called pulmonary hemorrhage is actually the most severe manifestation of pulmonary edema rather than vascular disruption.35 This distinction has been drawn by measuring the hematocrit of hemorrhagic fluid suctioned from the airway.36 The airway fluid generally will be 15 to 20 points lower than whole blood in the same patient at that time. Finding whole blood in the airway is rare and is usually a result of trauma from suctioning.
Similar to older children and adults, factors that alter Starling forces within the pulmonary microcirculation will predispose the lung to hemorrhagic pulmonary edema. These factors include increased perfusion pressure (e.g., left ventricular failure), increased blood flow (e.g., from a left-to-right shunt across a patent ductus arteriosus [PDA]), increased microvascular permeability (e.g., associated with sepsis or oxygen toxicity), and decreased oncotic pressure (e.g., protein malnutrition or water overload). Most infants who have pulmonary hemorrhage will have more than one risk factor present. In the neonate, the factors most commonly associated with hemorrhagic pulmonary edema are those that increase pulmonary blood flow, such as a left-to-right shunt or treatment with surfactants.35,37 Clinical features that increase the risk of hemorrhagic pulmonary edema or even frank pulmonary hemorrhage in neonates include extreme preterm gestation, asphyxia, hypothermia, certain types of congenital heart disease, and underlying coagulopathies.38 Pulmonary hemorrhage also has been associated with neurologic disorders, including seizures, stroke, subarachnoid hemorrhage, and massive intraventricular hemorrhage. This association has led some researchers to suggest that pulmonary hemorrhage may have a neurogenic origin and that neonates with known or suspected neurologic injury represent a particularly high risk group.39
Although the diagnosis of hemorrhagic pulmonary edema is relatively straightforward, management can be challenging. The most important approach to managing hemorrhagic pulmonary edema is to establish high positive end-expiratory pressure (PEEP); this not only effectively reduces alveolar flooding but also improves oxygenation and left ventricular function. Although the airway must be kept clear, frequent suctioning not only may be traumatic but also can aggravate the condition by reducing PEEP. High mean airway pressures, which can be safely achieved with HFV, can be effective in massive pulmonary hemorrhage with rapid improvement in oxygenation.40
Some persons have advocated administration of epinephrine or iced saline solution via the endotracheal tube, but the efficacy of this method is questionable, and epinephrine may worsen the condition by elevating pulmonary vascular pressures. Aggressive volume resuscitation also should be avoided for the same reason. Antibiotics should be considered if sepsis is clinically suspected, but the efficacy of prophylactic antibiotics to prevent bacterial contamination of the airways is unproven. If an underlying cause such as a PDA or coagulopathy is suspected, it should be treated accordingly. Additional therapies under investigation include surfactant41 and recombinant activated factor VII.42 Prevention also may be possible in some cases; in preterm infants at high risk of the development of hemodynamically significant PDA, indomethacin prophylaxis appears to reduce the incidence of severe hemorrhagic pulmonary edema.43 In a recent randomized trial of hemocoagulase given to preterm newborns requiring mechanical ventilation, the incidence of pulmonary hemorrhage (suspected or proven) was reduced by 70%44; however, in that study, the definition of “pulmonary hemorrhage” was rather broad and occurred in more than 40% of control infants, a much higher rate than is typically reported for this condition.
Even with aggressive management, the incidence of mortality from hemorrhagic pulmonary edema can exceed 25%, and surviving infants are at increased risk for neurodevelopmental impairment (e.g., cerebral palsy or cognitive delay) and neurosensory impairment (e.g., hearing loss requiring amplification or bilateral blindness).43
Pneumonia
The lungs represent the most commonly affected organ in neonates with sepsis. Bacterial or viral infection of the neonate may begin in utero, either by transplacental passage or, more commonly, ascending infection from the maternal genital tract. A delay from the time of rupture of the amniotic membranes until delivery increases the risk of an ascending infection, although some organisms may invade through intact membranes. Cervical bacterial colonization with group B streptococci or primary herpesviral cervical infection during pregnancy increases the risk of transmitting those diseases; however, routine cervical cultures taken during pregnancy often do not reliably predict the actual flora at the time of delivery. Also, infants born vaginally are invariably colonized with organisms from the vaginal canal and typically swallow organisms during vaginal passage. Cesarean delivery is not necessarily protective because fetuses may swallow contaminated amniotic fluid or aspirate organisms in utero. Infection occurring during the perinatal period may not present clinically for several days; thus infections congenitally acquired may be indistinguishable from infections postnatally acquired (i.e., nosocomial).
Organisms that cause perinatal pneumonias, then, typically are those found in the genital tract of the mother and include streptococci (groups A, B, and D), gram-negative rods (e.g., Escherichia coli and Klebsiella species), Listeria monocytogenes, Ureaplasma, genital hemophilus, and herpesvirus. Less commonly, maternal viral infections due to adenovirus, enteroviruses, or varicella can be vertically transmitted to the fetus. Although perinatal tuberculosis is rare compared with other causes of neonatal pneumonia, the increasing prevalence of this disease in women of child-bearing age increases the likelihood of new cases45; in congenitally acquired cases, the mother may be symptom free.46 Perinatal infection with other organisms, most typically chlamydia, may not present for several weeks.
Pneumonia as a nosocomial infection may develop in neonates, particularly those who require mechanical ventilation for other critical illness. Although reported rates vary widely, in part because of the lack of a diagnostic gold standard in this population, some authors have suggested that the incidence of ventilator-associated pneumonia may be as high as 30% in selected NICU populations.47,48 In addition to the presence of an endotracheal tube, risk factors for ventilator-associated pneumonias include low birth weight, prolonged mechanical ventilation, sedation with opiates, frequent endotracheal tube suctioning, and the crowding that is typical in some units.47,49 If a ventilator-associated pneumonia is suspected, typical nosocomial pathogens such as staphylococcus, Klebsiella and Pseudomonas species, and the pathogens previously listed for congenital pneumonias should be considered as possible causes.
Congenitally acquired pneumonia/sepsis can be a rapidly fatal disease, especially in the case of group B streptococcal or herpesviral infections, for which mortality rates as high as 50% have been reported.50,51 A high degree of suspicion is therefore important because prompt treatment may be lifesaving. Although antibiotics are routinely used in neonates with suspected pneumonia or sepsis, antiviral therapy should be considered if the infant has systemic signs such as shock or disseminated intravascular coagulation or is not responding to initial therapy.
In regard to group B streptococcus (GBS) infection, guidelines for antenatal screening and antepartum prophylaxis published by the Centers for Disease Control and Prevention in 1996, and revised in 2002, have been followed by a significant decrease in GBS-related neonatal morbidity and mortality in the United States.52 However, because of the inability of current microbiologic screening to identify all carriers, the failure in some cases to administer adequate intrapartum prophylaxis, and the need to deliver some infants prior to scheduled screening, GBS continues to be an important cause of early onset sepsis.53 Given the complications and potential limitations associated with maternal screening and intrapartum prophylaxis, vaccines may be the most effective means of preventing neonatal GBS disease and are currently under development.53
Meconium Aspiration Syndrome
Passage of meconium in utero is generally considered a sign of fetal distress. The stress can be acute, as in the case of cord compression during labor, or chronic, as in the case of preeclampsia. Moderate distress occurring during labor results in passage of meconium during the final stages of delivery (terminal meconium), whereas more severe or chronic distress results in passage in utero, with resultant staining of the amniotic fluid and fetus. The incidence of meconium staining as a more significant marker of fetal distress occurs in 10% to 20% of all deliveries and is most common in postmature infants.54 There also appears to be a maturational aspect to the ability to pass meconium because it is rarely observed in fetuses younger than 36 weeks’ gestation.54
Not only is meconium-stained amniotic fluid a sign of antenatal distress, but it also can cause subsequent difficulties in the neonate. The contaminated amniotic fluid may be aspirated by the fetus, either in utero or during passage through the birth canal, and lead to subsequent respiratory distress. Meconium is a lipid and protein-rich substance that is highly irritating to mucous membranes of the distal airways, resulting in a chemical pneumonitis.55 Dissolved meconium may travel down the respiratory tree and inactivate pulmonary surfactant; this inactivation leads to a functional surfactant deficiency.56,57 In addition, meconium induces a potent inflammatory response, further impairing lung function.58,59 As inflammatory markers improve, so too does pulmonary function.60 More particulate meconium will remain trapped in small airways, and this leads to a ball-valve type of gas trapping. In most cases the meconium is gradually removed from the respiratory tract through phagocytosis, and normal pulmonary function returns in 5 to 7 days. In more severe cases, meconium aspiration syndrome may lead to respiratory failure, and even death, despite aggressive intervention.
Infants with meconium aspiration are typically postmature, with elongated nails, peeling skin, and staining of the umbilical cord, skin, and nails. Respiratory distress develops soon after birth, although the infant’s respirations initially may be depressed if meconium passage occurred in response to a recent asphyxial episode in utero. Gas trapping may lead to a barrel-shaped appearance to the chest, and signs of respiratory distress may be severe. Chest radiographs often show characteristic patchy densities, hyperinflation, and areas of collapse. Air leaks are especially common. Aspiration of blood during delivery results in a similar clinical and radiographic picture; however, blood aspiration usually has a much milder course.61
An important step in the treatment of meconium aspiration syndrome is prevention. This has led to the routine practice of aggressive suctioning, beginning with clearing of the infant’s nose and mouth while still at the perineum and before delivery of the infant’s chest, followed by endotracheal intubation once the infant is delivered. A couple of large, randomized clinical trials have brought these practices into question and have even raised concerns that such interventions may produce harm, particularly in the vigorous infant.62,63 On the basis of these trials, current recommendations are that suctioning should be limited to infants who are not vigorous at delivery.64 Another intervention aimed at reducing disease as a result of meconium aspiration has been amnioinfusion, that is, the introduction of fluid transcervically during labor. The theoretical benefits of this approach include dilution of thick meconium and reducing cord compression by providing support to the umbilical cord. However, a recent large, randomized clinical trial has put this practice into question.65 As with tracheal suctioning, it can be argued that distressed fetuses may aspirate in utero, long before delivery, and that severe complications such as persistent pulmonary hypertension are more likely the result of the antecedent stress and are not due to meconium aspiration per se.
The treatment of meconium aspiration syndrome is to provide supportive care; infants are recognized as being at increased risk for having persistent pulmonary hypertension (see section on “Persistent Pulmonary Hypertension of the Neonate”). Supplemental oxygen support to maintain arterial oxygen saturation, endotracheal suctioning to clear remaining meconium, and ventilatory techniques to minimize gas trapping are techniques that are commonly used. HFV may be helpful in preventing subsequent air leaks. Antibiotics are commonly used because distinguishing the clinical and radiographic picture from sepsis may be difficult and because damage to the airways may predispose to subsequent bacterial infection. Surfactant therapy has been recently explored as an adjunctive therapy, following promising results from two small pilot studies.66,67 A recent systematic review of four clinical trials in term and near-term infants concluded that surfactant administration significantly reduced the need for extracorporeal life support, although the overall incidence of mortality was not affected68; in many centers, surfactant replacement has been added to other therapies routinely use in this condition, including high-frequency ventilation and inhaled nitric oxide.
A recent modification of surfactant replacement has been to lavage the lung with relatively large volumes of diluted surfactant to facilitate removal of meconium and improve surfactant function.69,70 While this approach has merit, more clinical work needs to be done to determine the optimal approach, because some infants do not tolerate the procedure.70
Despite the intense inflammatory nature of meconium aspiration, the utility of steroid administration remains unclear54; although a recent small, randomized, controlled trial showed some clinical improvements, the mortality rate remained high.71
Because meconium passage in utero and meconium aspiration syndrome per se often are associated with an hypoxic event, long-term outcome remains guarded, particularly with regard to neurodevelopment.72 In addition, a recent study has suggested that meconium aspiration during the perinatal period may be associated with increased risk of reactive airway disease in early childhood.73
Surfactant-Deficient Respiratory Distress Syndrome
Pulmonary surfactant disperses at the air-liquid interface on the inner surface of the alveolus, reduces surface tension at that interface, and prevents alveolar collapse at end-expiration. Surfactant is produced by type II alveolar epithelial cells and is a phospholipid and glycoprotein composite with phospholipid as the surface-active agent and glycoproteins aiding in surface adsorption, spreading, and metabolism of surfactant.74 Maturation of the pulmonary surfactant system is generally not complete until the latter part of the third trimester of fetal life, but it can be induced by intrauterine stress, by maternal steroid therapy, and after preterm delivery. The incidence of surfactant deficiency at birth is inversely related to gestational age; approximately 50% of infants born between 30 and 36 weeks’ gestation will be surfactant deficient, and virtually all infants born before 28 weeks’ gestation will be affected to some degree.75
Surfactant-deficient alveoli are more prone to collapse, and this leads to diffuse atelectasis, reduced ventilatory compliance, and intrapulmonary shunting. Infants with surfactant deficiency have stiff, noncompliant lungs and require significant distending pressure for the lungs to be ventilated. Neonates will have tachypnea, retractions, and expiratory grunting; these symptoms indicate RDS. Preterm infants with RDS have significant morbidity, although some complications may primarily be a result of prematurity per se. Short-term complications can be life-threatening and include pulmonary air leaks, pulmonary hemorrhage, and intracranial hemorrhage. Infants with RDS are at risk for later complications including necrotizing enterocolitis and chronic lung disease. Infants who require prolonged intubation and mechanical ventilation also are at risk for subglottic injury including subglottic stenosis and tracheomalacia. Before surfactant therapy, the mortality rate from RDS exceeded 20%76; now infants rarely succumb to RDS unless severe complications develop.77–79
Assessment of fetal lung maturity using phospholipid analysis of amniotic fluid and maternal steroid therapy, both introduced in the mid 1970s, have reduced the incidence of RDS in infants born before term. Treatment with surfactants in neonates with symptoms during the first few days of life has significantly reduced the clinical severity of RDS and improved survival.24
Unfortunately, although surfactant therapy has reduced some short-term complications such as pulmonary air leaks, the incidence of long-term morbidity remains unchanged, in part because of the increased survival rate of preterm infants with extremely low birth weight (<1000 g).78,79
Surfactant Protein B Deficiency
Pulmonary surfactant consists of surface-active phospholipid and a small amount (approximately 10% by weight) of protein.74 Various surfactant-associated proteins (SPs) have been identified. SP-A and SP-D are large molecular weight, hydrophilic glycoproteins. Although these larger proteins have no surface tension–lowering ability per se, SP-A regulates surfactant phospholipid synthesis, secretion, and recycling and blocks the inhibition of native surfactant by plasma proteins that may leak into the alveolus during lung injury. SP-D does not appear to be involved in surfactant function but does play a key role in host defense of the lung. Despite having little to do with surfactant function, a recent study found that certain SP haplotypes for SP-A and SP-D conferred protection against newborn RDS.80 In contrast to SP-A and SP-D, SP-B and SP-C are small, hydrophobic glycoproteins that promote spreading of surfactant across an air-liquid interface, which is an essential prerequisite for surfactant to function. A deficiency of either SP-B or SP-C markedly interferes with natural surfactant function in animal models.74
Congenital alveolar proteinosis is a rare disease entity with histopathologic similarities to alveolar proteinosis in older children and adults.81 The clinical course of congenital alveolar proteinosis, however, is markedly different, characterized by rapid progression to death within several hours to days. A clustering of cases within families has suggested a genetic basis for this disorder; the condition in older children and adults is thought to result from a nonspecific alveolar injury. Pulmonary lavage has been helpful in adults and some children with alveolar proteinosis, but it has not been studied in infants with congenital disease.82 Extracorporeal support has not altered the long-term prognosis of infants with this condition.83
In the early 1990s, several cases of congenital alveolar proteinosis were described in which SP-B and its messenger protein have been shown to be totally absent.84,85 Since then, more than 25 loss of function mutations have been identified in patients with SP-B deficiency, the most common being a single gene mutation (121 ins2).86 Adult humans heterozygous for this mutation have normal pulmonary function,87 whereas homozygous cases are uniformly lethal within days of life. Affected infants respond only transiently to exogenous surfactants that contain SP-B88; to date, the only survivors are those who have undergone a lung transplant. Molecular techniques can identify affected infants and even predict fetal outcome, allowing us to provide specific counseling for parents whose infants have this fatal disorder.89
Congenital Malformations of the Lung
Pulmonary Hypoplasia
Both static and dynamic expansion of the fetal lung appears to be an important determinant of normal fetal lung development.90 Static lung expansion occurs as a result of fetal lung liquid production. Epithelial cells within the lung actively secrete fluid into the lung lumen, distending the future airspaces. An intraluminal pressure gradient above amniotic fluid pressure is maintained by glottic regulation of fluid efflux from the trachea into the amniotic cavity, thus keeping the lungs expanded at a fluid volume that approximates postnatal functional residual capacity.91 Failure to maintain this distention, either by inadequate production or excessive drainage of fetal lung liquid, leads to developmental hypoplasia. Dynamic lung expansion occurs during fetal breathing movements, which are rhythmic in nature and occur with increasing frequency during the latter part of gestation.92 Absent or abnormal fetal breathing also appears to result in pulmonary hypoplasia.90
Pulmonary hypoplasia, unlike pulmonary agenesis or aplasia (discussed later in this chapter), can occur any time during gestation. Hypoplastic lungs are small in volume, and DNA content relative to body size have reduced numbers of alveoli, bronchioles, and arterioles per unit mass.90 Although the pathophysiologic origin is not well understood, pulmonary hypoplasia is thought to result from impairment of normal fetal lung expansion and generally occurs in conjunction with one of the following conditions: (1) space-occupying lesions within the hemithorax, such as a diaphragmatic hernia, or massive pleural effusions associated with fetal hydrops; (2) an inadequate thoracic cage, as in asphyxiating thoracic dystrophy or achondrogenesis; (3) a deficiency of amniotic fluid (oligohydramnios), either because of leakage (e.g., preterm rupture of fetal membranes) or underproduction (e.g., renal dysplasia) 25; (4) inadequate vascular supply to the developing lung, as may be seen with pulmonary artery atresia, hypoplastic right heart, or tetralogy of Fallot; (5) lack of the fetal breathing movements that normally occur throughout the latter part of gestation; and (6) chromosomal anomalies such as trisomy 13 or 18. Pulmonary hypoplasia may occur in the absence of any of these conditions, but such cases of primary isolated pulmonary hypoplasia are rare.90
Infants with pulmonary hypoplasia generally have signs of respiratory failure in the immediate newborn period. Reduced lung volumes impair ventilation and lead to hypercarbia, and decreased surface area for gas exchange (due to decreased alveoli) leads to hypoxemia. A decreased cross-sectional area of the vasculature makes these infants particularly susceptible to pulmonary hypertension, which further exacerbates the hypoxemia. The chest radiograph in infants with pulmonary hypoplasia should show low lung volumes but may otherwise be unremarkable. The severity of the respiratory distress depends on the degree of hypoplasia and the presence of associated problems such as fetal hydrops or cyanotic heart disease. The most common association is renal dysplasia or agenesis; in these cases infants have a history of moderate to severe oligohydramnios and have severe respiratory distress and compression deformities of the face and extremities (Potter’s syndrome).93
Congenital Diaphragmatic Hernia
Failure of the pleuroperitoneal canal to close at 6 to 8 weeks’ gestation results in a diaphragmatic defect that allows gastrointestinal structures to travel into the thoracic cavity as the intestines return from outside the fetus to the abdominal cavity.94 The resulting mass effect in the chest exerts a negative influence on ipsilateral lung growth, characterized by a quantitative reduction in airways and their associated preacinar arteries. Congenital diaphragmatic hernia (CDH) occurs in approximately 1 in 3000 births and is the most common cause of pulmonary hypoplasia in the neonate.95 The defect occurs on the left side in 80% to 85% of cases; the reason is that closure of the right pleuroperitoneal membrane normally precedes the left during fetal development. Because herniation often occurs before the tenth week of gestation when normal gut rotation occurs, malrotation is common. Nongastrointestinal anomalies are found in approximately 25% of cases; the most common involve the cardiovascular system, where virtually any kind of defect has been reported.94–96 Other associated anomalies include esophageal atresia, trisomies (13, 18, and 21), Turner’s syndrome, neural tube defects, and renal anomalies. While it has been assumed the pulmonary hypoplasia seen with CDH is secondary to mechanical forces, studies in newborns and animals have found that CDH may be associated with nutritional deficiencies.97,98 In the rat model of nitrofen-induced CDH, prenatal retinoic acid improves alveologenesis99; the experimental data regarding vitamin E supplementation has yielded conflicting results.100–102
Clinical presentation of CDH depends on the degree of pulmonary hypoplasia present (see section on “Pulmonary Hypoplasia”). In addition, the abdomen is often scaphoid because of a paucity of abdominal contents. As the infant cries and swallows air, the degree of lung compression may worsen, and an infant who appears healthy at delivery may undergo decompensation within minutes. The chest radiograph will show a cystic lesion in the lower lung field, often extending upward along the lateral chest wall. Initially, while the intestines remain fluid filled, the radiograph may be similar to that seen with pulmonary sequestrations or fluid-filled cysts (discussed later in this chapter); as the infant swallows more air, the radiographic findings can be confused with congenital emphysema or even a pneumothorax. Small or right-sided defects in infants may not present for weeks or even months; indeed, occasional cases have been diagnosed incidentally during childhood when chest radiographs are obtained for other reasons. Today, with the widespread use of antenatal sonography, most cases are diagnosed before birth, and significant confusion is avoided in the delivery room. In cases where sonographic findings are equivocal, prenatal magnetic resonance imaging may be particularly useful.103
Initial management focuses on stabilization, including immediate intubation and gastrointestinal decompression. Ventilation by bag and mask should be avoided because this intervention will only introduce more gas into the gastrointestinal tract. As with pulmonary hypoplasia, the clinical course is usually complicated by persistent pulmonary hypertension, which has accounted for mortality rates as high as 80%.104 With the advent of ECMO this number has dropped somewhat but is still significant.105 High-frequency ventilation appears to have particular merit for this condition and may improve mortality rates independent of bypass technology.96,106 In a lamb model of CDH the contralateral nonhypoplastic lung is functionally immature, leading some investigators to suggest that surfactant therapy may be beneficial,107 but this suggestion has not been borne out in a recent review of a large, national CDH registry.108 Attempts at intrauterine intervention, either to close the defect or to encourage lung growth through temporary blocking of the fetal lung liquid egress at the trachea, have been disappointing; fetal surgery is associated with an unacceptably high incidence of complications, including recurrence of the defect, preterm delivery, and miscarriage.96,109 In infants with significant pulmonary hypoplasia who require ECMO, distending the lung with perfluorochemical can promote lung growth, but further study is needed.110
Although early corrective surgery had been advocated in the past, recently a paradigm shift toward delayed repair has occurred, in large part because of the observation that respiratory function often worsens in the immediate postoperative period.95,96,111,112 Thus early, aggressive cardiorespiratory stabilization followed by surgery has become the recommended approach and is associated with improved outcome.95,113 An alternative to an initial period of attempted stabilization has been an ex-utero intrapartum therapy procedure, in which the delivering fetus is orally intubated and placed on a mechanical ventilator prior to umbilical cord ligation. In this approach, a brief trial of ventilation may be given; if oxygen saturation does not improve during the trial, the fetus is cannulated and ECMO is begun, followed by delivery of the infant. This approach may be helpful in selecting infants who are least likely to respond to initial stabilization in the NICU, and reported outcomes with this approach have been favorable, although the mortality rate for infants with this condition remains quite high.114
Cystic Adenomatoid Malformation
Cystic adenomatoid malformation (CAM) is a relatively infrequent lesion, estimated at 1 in 30,000 pregnancies.115 It results from abnormal mesenchymal proliferation and failure of maturation of bronchiolar structures early in gestation.116,117 The resultant adenomatous overgrowth leads to the development of cysts and suppression of alveolar growth. The cysts are almost always multiple, and in more than 95% of cases the cystic malformations lie within a single lobe. No lobar predilection exists. Histologically, the lesions are notable for the preponderance of elastic tissue and for a lack of cartilage. The cysts communicate directly with the tracheobronchial tree and with each other.
CAMs have been divided into three types, which vary both in anatomic and clinical characteristics. According to the traditional Stocker classification scheme, Type 1 CAM, which accounts for about half the cases, occurs as a few large (>2 cm) cysts, usually one to four in number, or a single large cyst surrounded by much smaller “satellite” cysts. Type 2 CAM, which accounts for about 40% to 45% of the cases, consists of multiple, small (<2 cm), evenly spaced cysts scattered throughout the affected area. Compared with type 1 CAM, type 2 cysts are associated with a much higher incidence (about 25%) of anomalies in other organs, particularly within the genitourinary tract (e.g., renal dysgenesis). Type 3 CAM, which accounts for less than 10% of cases, occurs as large collections of numerous tiny cysts; the affected area can be large, and this type often leads to early cardiovascular compromise, resulting in fetal hydrops or immediate postnatal complications. The Stocker classification scheme is based primarily on postnatal lung examinations; recent studies of fetal lung specimens suggests that a different classification scheme may be needed, particularly as this condition is now detected antenatally in most cases.118,119
Treatment of infants with symptoms may require positive pressure ventilation; in some infants PEEP may facilitate emptying of the cysts. Definitive treatment is surgical removal of the affected lobe, which can be done thoracoscopically in many cases.120 Even if an infant is asymptomatic, surgical resection is recommended by 3 to 6 months of age because of the high risk of expansion or infection if the cysts are left untreated.115,121
Prognosis depends on the type and extent of the CAM. The large type 3 lesions are more likely to cause immediate distress and carry a higher incidence of mortality, especially if associated with pulmonary hypoplasia or fetal hydrops.115,121 The prognosis in type 2 CAM depends on the presence and nature of associated anomalies. In addition, malignant transformation of CAM has been reported.94,116,121 Most cases of CAM, however, carry a good prognosis.115,117,121
Bronchogenic Cysts
Bronchogenic cysts occur as a result of anomalous budding of the ventral or tracheal diverticulum of the foregut during the sixth week of gestation, with subsequent separation from the normally developing bronchi by the sixteenth week of gestation.116,117 If separation occurs early (<12 weeks), the bronchogenic cyst tends to be located in the mediastinum (which is the most common type); if separation occurs later, it is more likely to occur in the peripheral pulmonary parenchyma. The cyst walls are cartilaginous and receive either systemic or pulmonary blood supply depending on their location. Bronchogenic cysts are more common in male infants, are usually singular, are more commonly right sided, and are generally less than 10 cm in diameter. They generally do not communicate with the airway and remain fluid filled, which differentiates them from pulmonary parenchymal cysts.
Chest radiographs can readily disclose most bronchogenic cysts.122 The cyst typically appears as a round or oval water-density mass, commonly in the mediastinal or perihilar area; if the cyst has been infected, an air-fluid level may be present. Mediastinal bronchogenic cysts usually appear just beneath the carina and extend to the right. Pulmonary bronchogenic cysts are usually sharply circumscribed and appear toward the periphery; two thirds of these cysts will be located in the lower lobes, with no right or left predilection. About 25% of bronchogenic cysts may be difficult to visualize with a chest radiograph; in these cases CT scans cannot only delineate the lesion but can also discern associated anomalies, such as a pulmonary sequestration (see later).116,122
Treatment may require ventilatory support for infants with symptoms. In all cases, surgical resection is indicated. Prognosis for infants with bronchogenic cysts, whether mediastinal or pulmonary, is good.123
Pulmonary Parenchymal Cysts
Pulmonary parenchymal cysts are thought to represent a disorder of bronchial growth,116,117 although they also may be acquired.124 Like adenomatoid malformations and bronchogenic cysts, congenital cysts arise early in fetal life; pulmonary parenchymal cysts are thought to develop at a time when completion of the terminal bronchioles and development of the alveoli are occurring. Pulmonary cysts are typically thin walled, singular, multilocular, and located in the periphery. Unlike bronchogenic cysts, some communication usually exists between the pulmonary cyst and the tracheobronchial tree, and thus approximately 75% will become filled with air. Like adenomatoid malformations, pulmonary cysts contain mostly elastic tissue and little or no cartilage.
Although pulmonary cysts are generally small (1 to 2 cm in diameter), they can expand dramatically and thus are much more likely to be symptomatic than are bronchogenic cysts. As with adenomatoid malformations, the lack of cartilaginous support leads to trapping of air. Unlike adenomatoid malformations, pulmonary cysts are rarely associated with other anomalies. Rupture of a peripheral cyst can result in a pneumothorax. Rarely, multiple cysts can occur and involve both lungs in an extensive fashion; these cases are generally fatal within the perinatal period.
Chest radiographs typically reveal thin-walled, round cysts with an air density. Often faint strands of lung tissue can be seen within the cysts. A large pulmonary cyst may be confused with congenital lobar emphysema; in this case a CT scan should easily distinguish the cystic nature of the former condition.122
Reports of spontaneous resolution of pulmonary cysts have been infrequent. As with other cystic lesions of the lung, however, surgical resection of the affected lobe is usually indicated.116,124
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


