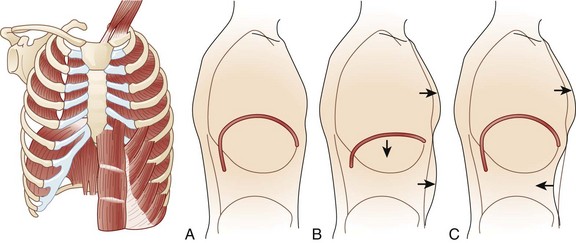
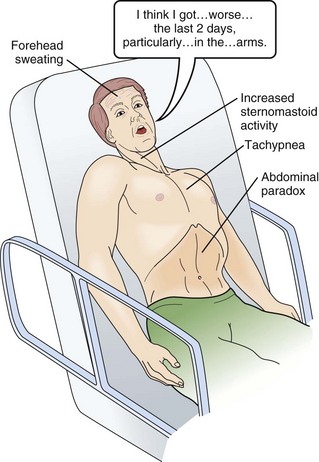
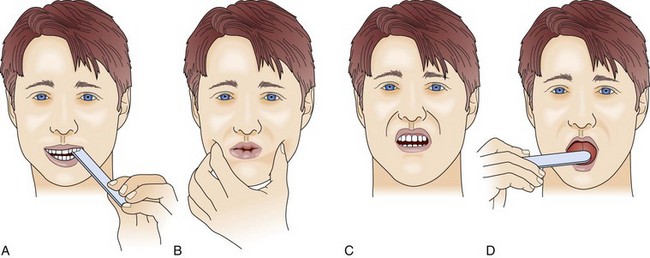
64 MG is an autoimmune disease of defective neurotransmission leading to fatigable muscle weakness. The incidence is 0.5 to 5 cases per 100,000. Its pathogenesis can be summarized briefly as an antibody reaction at the antigen epitopes of the acetylcholine receptor (AChR), eventually leading to destruction and simplification of the junctional fold and widening synaptic cleft.1 Onset may be at any age but tends to be earlier in women (mean age 28 years) than in men (mean age 42 years). MG results in either transient or persistent focal or generalized fatigability or weakness. In more than half of the cases, the initial symptoms involve the eyes, presenting as ptosis, diplopia, or ophthalmoparesis. Myasthenic crisis is arbitrarily defined as MG complicated by respiratory failure requiring mechanical ventilation or delayed extubation for more than 24 hours in an already intubated myasthenic patient. It is seen in 10% to 60% of MG patients typically within the first 12 months of disease onset.2,3 The need for ventilatory assistance usually follows onset of weakness of diaphragmatic or accessory respiratory muscles, but mechanical ventilation also may become necessary because of airway collapse from oropharyngeal muscle weakness, stridor from vocal cord weakness, or the inability to clear secretions.4 One third of the patients may have recurrent myasthenic crises.5 Common precipitating causes include viral or bacterial infection in 48% (pneumonia, respiratory infection, or sepsis), aspiration in 10%, reduction of pyridostigmine in 19%, and initiation of steroids in 7% patients.6,7 Changes in medication may relate to the recent addition of corticosteroid, dose reduction, or high-dose acetylcholinesterase inhibitors. Other precipitating factors may be physical trauma, surgical procedures (particularly thyroidectomy), pregnancy, emotional stress, or exposure to drugs with neuromuscular blocking action such as aminoglycosides. No precipitating factor can be identified for myasthenic crisis in 30% patients.6,7 Myasthenic crisis requires urgent evaluation and treatment. The mortality rate for myasthenic crisis has declined from a nearly always fatal outcome in the 1920s to 4.5% in the new millennium with the discovery and application of acetylcholinesterase compounds, mechanical ventilation, immunosuppressive agents, and more recently, plasma exchange and intravenous immunoglobulin (IVIG).8 Timely diagnosis and appropriate treatment with IVIG or plasma exchange for patients with bulbar and respiratory muscle weakness have helped to reduce the mortality rate and total hospital stay from myasthenic crisis, but care of a patient with MG may be prolonged and complicated. Median hospital stay after a myasthenic crisis is 4 to 6 weeks. GBS is an acute, monophasic, inflammatory, demyelinating polyneuropathy, also known as acute inflammatory demyelinating polyneuropathy (AIDP). Its incidence is 1 to 2 cases per 100,000 people and remains fairly constant across the continents. GBS commonly is precipitated by an infection, but the immunopathogenesis has remained elusive since its original description in 1916. Campylobacter jejuni, cytomegalovirus, and Epstein-Barr virus predominate as preceding infection pathogens.9 Symptoms and signs usually progress within 1 to 2 weeks. The diagnosis of GBS often is straightforward. Severe back pain and limb paresthesias, starting in the ankles and wrists with a “tight band” feeling, are typical presenting signs. The paresthesias gradually scatter over the limbs and move proximally. Although paresthesias are often the presenting symptoms, sensory modalities remain normal to mildly impaired. Weakness begins in the more proximal muscles, causing difficulty with climbing stairs and getting out of a chair, and is notable 1 or 2 days after the onset of paresthesias. Symmetrical weak muscles are accompanied by depressed or absent deep tendon reflexes. Mostly legs are more involved than arms, creating the impression of an ascending paralysis. Facial and oropharyngeal muscles are affected in 50% of the cases, and weakness of these muscle groups may be the initial manifestation. Patients may also have staccato speech (ability to speak only short sentences) and small tidal volume with increased respiratory rate. Respiratory failure, if it occurs, commonly appears within 1 week after the onset of paresthesias.10 Dysautonomia occurs in up to 70% of the cases, manifesting as arrhythmias, tachycardia, diaphoresis, labile blood pressures, urinary retention, or ileus. Cerebrospinal fluid (CSF) analysis typically shows high protein content with a normal white blood cell count (the classic albuminocytologic dissociation). More than 10 white blood cells may be seen although this number is unusual but is more common in associated disorders such as Lyme disease, sarcoidosis, and AIDS.11 Electrophysiologic studies are more useful for diagnosis and less useful for prognostication. The typical finding involves demyelination seen as conduction block or increased conduction velocities. Specific tests for proximal nerve involvement seen early in the disease course include recording of F waves and nerve signal abnormalities like dispersion or dropout in signal. Prolonged F wave may be the only finding early on. Needle electromyography may be normal in the first 2 weeks. Recovery often starts after the second week. Progression over 8 weeks more likely suggests the diagnosis to chronic inflammatory demyelinating polyneuropathy (CIDP). During respiration, lungs can expand and recoil in two ways: by downward and upward movement of the diaphragm to lengthen and shorten the chest cavity, and by elevation and depression of the ribs to increase and decrease the anteroposterior diameter of the chest (Fig. 64.1). Normal quiet breathing is largely accomplished by contraction of the diaphragm. In neuromuscular respiratory failure, ventilatory function is compromised through two mechanisms: (1) respiratory muscle weakness or fatigue (involving the diaphragm and the intercostal muscles), and (2) oropharyngeal weakness, which leads to obstruction of the upper airway and inability to clear secretions. The neuromuscular respiratory failure follows the following pattern: failure of diaphragm and intercostal muscles followed by use of accessory muscles resulting in hypoventilation and atelectasis, further leading to shunting and hypoxia. Respiratory muscle weakness leads to shallow low tidal volume breathing and poor gas exchange leading to tachypnea and later hypercapnia. Oropharyngeal muscle weakness leads to aspiration pneumonitis and could further worsens the hypoxia. Clinically, the patient will pause frequently during speech and breathlessness improves in an upright position (Fig. 64.2). The first indication of diaphragmatic weakness is alveolar hypoventilation and impaired CO2 exchange. These changes are followed by an increase in respiratory rate as a compensatory mechanism to attempt to maintain minute ventilation. Later, accessory muscles of ventilation are recruited in response to increased ventilatory demand. Paradoxical breathing, also known as thoracoabdominal asynchrony, occurs with severe respiratory weakness. Normally, the abdomen and chest expand and contract in a synchronized fashion. During inspiration, a downward movement of the diaphragm pushes the abdominal contents down and out as the rib margins are lifted and moved out, causing both chest and abdomen to rise. With diaphragmatic weakness or paralysis, the diaphragm moves up rather than down during inspiration, and the abdomen moves in, contracting during chest rise. This is known as “paradoxical breathing” and can be visualized with fluoroscopy (see Fig. 64.1). The ventilatory drive response to an increase in CO2 in patients with GBS and MG has been studied and was found to be unlikely to contribute to hypoventilation during ventilatory failure.12 Ventilatory drive increases during acute hypoventilation, and the ventilatory drive response to CO2 remains intact, even when the minute ventilation response to CO2 is poor. Although the upper airway muscles do not contribute directly to chest expansion or collapse, they are essential for keeping the airways open during respiration. They play an important role in preventing collapse of the pharynx during inspiration and preventing aspiration during swallowing. With the exception of laryngeal muscles, oropharyngeal muscles have a higher proportion of fast fibers. Their weakness can be seen early on in MG and later in GBS (Fig. 64.3). Slow fibers have a higher fatigue resistance than fast fibers due to their highly oxidative metabolism. The diaphragm has an equal proportion of slow and fast muscle fibers, which in association with small fiber size, high aerobic oxidative enzyme activity, and large number of capillaries make it resistant to fatigue.13 Frequent (at least four times daily) assessment of pulmonary function should be instituted using bedside spirometry. Although bedside spirometry is often done for respiratory evaluation in both MG and GBS, it is less reliable for MG as patients’ results often vary depending on their activity level prior to the testing. Patients should be coached for spirometry as they may have difficulty making an adequate seal around the mouthpiece of the spirometer. Poor effort and leak around the mouthpiece may lead to lower vital capacity (VC), maximal inspiratory pressure (PImax), and maximal expiratory pressure (PEmax) than expected based on arterial blood gas (ABG) and chest x-ray studies. VC, the volume of exhaled air after maximal inspiration, is 60 to 70 mL/kg in normal persons, and is determined primarily by the size of the thorax and lungs. In neuromuscular respiratory failure, reduction of VC to 30 mL/kg is associated with weak cough, accumulation of oropharyngeal secretions, atelectasis, and hypoxemia. Another measure of respiratory muscle strength is the ability to generate negative pressure with inspiratory effort. In normal persons, respiratory muscles cause pleural and alveolar pressures to change by approximately 3 cm H2O during the breathing cycle. Maximal pressure generation can be determined by blocking the upper airway and recording mouth pressure changes during inspiratory effort. PImax measures the strength of the diaphragm and other muscles of inspiration and reflects the ability to maintain normal lung expansion and avoid atelectasis. A maximal PImax is −114 cm H2O in young men and −67 cm H2O in young women (normal, exceeding −70 cm H2O). PEmax measures strength of the muscles of expiration and correlates with strength of cough and ability to clear secretions from the airway12 (Box 64.1). PEmax averages 160 cm H2O in young men and 95 cm H2O in young women (normal, greater than 100 cm H2O). This means that the respiratory muscles are capable of generating more than 30 times the amount of force necessary for tidal breathing.14 Worsening bedside spirometry can identify MG and GBS patients requiring invasive or noninvasive mechanical ventilation (NMV). An easy way to remember bedside pulmonary indicators of respiratory failure in GBS is the so-called 20/30/40 rule, with VC less than 20 mL/kg, a PImax less negative than −30 cm H2O, and a PEmax less than 40 cm H2O.
Muscular Paralysis
Myasthenia Gravis and Guillaine-Barré Syndrome
Clinical Presentation
Myasthenia Gravis
Guillain-Barré Syndrome
Acute Neuromuscular Respiratory Failure
Pathophysiology
Clinical Evaluation
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Muscular Paralysis: Myasthenia Gravis and Guillaine-Barré Syndrome