Chapter 100 Molecular Foundations of Cellular Injury
Necrosis, Apoptosis, and Autophagy
Cell Death
Each day, the human body produces and eradicates 60 × 109 cells, a rate of cell death of nearly 1 million per second.1 Cells die by one of three mechanistically distinct processes: apoptosis, autophagy, or necrosis (Table 100-1).2,3 These three forms of cell death can be distinguished by both morphologic and molecular biologic criteria.
Table 100–1 Differences Between Necrosis and Apoptosis
| Autophagy | Necrosis | Apoptosis |
|---|---|---|
| Cell shrinkage | Cell and organelle swelling | Cell shrinkage |
| Double-membrane vesicles | Early loss of membrane | Preservation of membrane integrity until late |
| Organelle degradation |
Modified from Hotchkiss RS, Tinsley KW, Swanson PE, et al: Endothelial cell apoptosis in sepsis, Crit Care Med 30(5 suppl):S225–S228, 2002.
In necrosis, an overwhelming acute injury is followed by cell and organelle swelling, with early dissolution of the plasma membrane and subsequent cell lysis. As the cell bursts, its contents enter the interstitial space, leading to an accompanying inflammatory response to the toxic enzymes and proteases released. Morphologically, cells undergoing necrosis show swelling of the entire cell and its internal organelles. Deoxyribonucleic acid (DNA) fragmentation has no characteristic pattern and exhibits a random pattern on electrophoretic gel analysis. As an “accidental” form of death, necrosis has always been described as an uncontrolled process. However, more recent evidence suggests that necrosis may be more controlled than previously thought.3
In contrast, apoptosis (also known as programmed cell death type I or cell suicide) is a well-controlled, evolutionarily conserved process.4,5 Following an appropriate trigger, a cell’s apoptotic machinery leads to orderly death of the cell. This is characterized by cell and organelle shrinkage (pyknosis), nuclear fragmentation (karyorrhexis), and cytoplasmic blebbing with retention of plasma membrane integrity. Ultimately, there is fragmentation of the cell into small apoptotic bodies that are phagocytosed by neighboring cells. Orderly DNA fragmentation is identifiable on electrophoretic gels as a characteristic “ladder” pattern. Because cytosolic contents are not released into the interstitial space, there is no accompanying inflammatory response.
Apoptosis is critical to the existence of virtually all multicellular organisms and is involved in widely divergent physiologic processes, including embryonic development, maturation, immunity, repair, and cellular homeostasis. However, alterations in the balance between cellular proliferation and death have been proposed to be involved in the etiology of cancer and autoimmunity6,7 and excessive apoptosis has been noted in the origins of neurodegenerative disease, osteoarthritis, allograft infection, graft-versus-host disease, type 1 diabetes, and heart failure.1,8 Importantly, apoptosis is increasingly recognized as playing a role in critical illness, in both septic and noninfectious inflammatory states. Because of the importance of apoptosis in multiple disease states, a number of ongoing phase II and phase III trials targeting apoptotic pathways are currently ongoing.1,9
Autophagy (also known as programmed cell death type II), is the only cell death process thought to be reversible and functions to allow cells to degrade cytoplasmic contents, including dysfunctional intracellular components, for either recycling or removal.10 Autophagy is characterized by formation of autophagosomes or large double-membrane vesicles that engulf intracellular organelles. Autophagosomes subsequently are trafficked to the lysosomal system for degradation. In times of starvation or stress, the breakdown of intracellular components provides the cell with a much-needed energy source. Autophagy, much like apoptosis, has been shown to be involved in a diverse number of physiologic processes, including embryonic development and immunity.11 Furthermore, over the last 5 years increases in autophagy have been associated with several disease states, including sepsis, cancer, heart disease, and Alzheimer disease.10 Finally, autophagy has been closely linked to apoptosis by the fact that both forms of cell death use many of the same signaling mechanisms and are induced by overlapping stimuli.12
Pathways of Apoptosis and Autophagy
Apoptosis is initiated by two main pathways: a receptor-mediated pathway and a mitochondrial-mediated pathway (Figure 100-1).13–16 The receptor-mediated pathway can be activated by a number of ligands, including Fas and tumor necrosis factor (TNF)-α. The mitochondrial pathway can be activated by a number of different stimuli following DNA damage, including reactive oxygen species, radiation therapy, and chemotherapy. Within the mitochondrial pathway, the Bcl-2 family of molecules plays a critical role. This group of proteins includes more than 20 proapoptotic and antiapoptotic molecules, many of which physically interact with each other.1,15,17,18 Their primary function appears to be regulation of cytochrome c release from the mitochondria, with proapoptotic family members promoting and antiapoptotic family members suppressing its release. The prototypical antiapoptotic proteins are Bcl-2, Bcl-xL, and Mcl-1, whereas the prototypical proapoptotic family members are Bax and Bak.
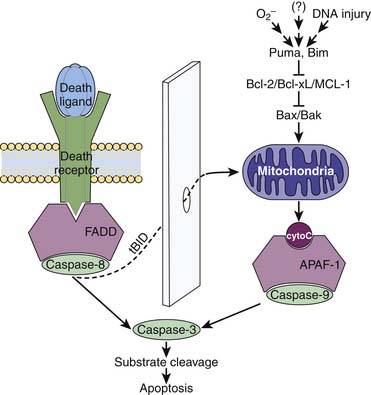
Both receptor-mediated and mitochondrial-mediated pathways activate specific members of the cysteine aspartyl-specific protease (caspase) family.19 Produced as inactive precursors, caspases are triggered by proteolytic processing as part of a cascade that produces their active counterparts. Depending on their location in the apoptosis cascade, caspases can be either upstream “initiators” or downstream “effectors” of cell death. Although several pathways for inducing these molecules exist, the receptor-mediated pathway acts by inducing caspase-8. In the mitochondrial pathway, cytochrome c binds to apoptotic protease-activating factor-1 (APAF-1), which in turn induces caspase-9. Both caspase-8 and caspase-9 converge on the final common pathway of apoptosis via induction of the death effector caspase-3, leading to the ultimate death of the cell.18 Crosstalk between the mitochondrial and receptor-mediated pathways of death can occur via the molecule Bid.
The pathways and regulation of autophagy are much less understood, but ongoing research is beginning to unravel some of the complex signals that lead to autophagy. Autophagy is known to occur at basal levels and is important in the maintenance of cellular homeostasis.10 Importantly, like apoptosis, autophagy can be a double-edged sword, in that too little or exaggerated levels can lead to unwanted consequences to the host. Key regulators of autophagy include ATG proteins and beclin-1, which are important in both formation of autophagosomes and autophagy signaling. It is important to note that many of the same genes that regulate apoptosis also regulate autophagy, the most studied of these being the Bcl-2 family and beclin-1. Antiapoptotic members of the Bcl-2 family inhibit beclin-1 function and proapoptotic family members disrupt this inhibition, thus promoting autophagy.20,21 Although a complete understanding of the relationship between these two forms of cellular death is not yet clear, there is growing evidence that both are important in a multitude of human diseases.
Human Studies
Several human studies have shown alterations in apoptosis, necrosis, and autophagy in critical illness. The potential functional importance of these alterations has led to the development of agents aimed at manipulating cell death for therapeutic benefit currently undergoing preclinical and clinical trials.1,9
Sepsis
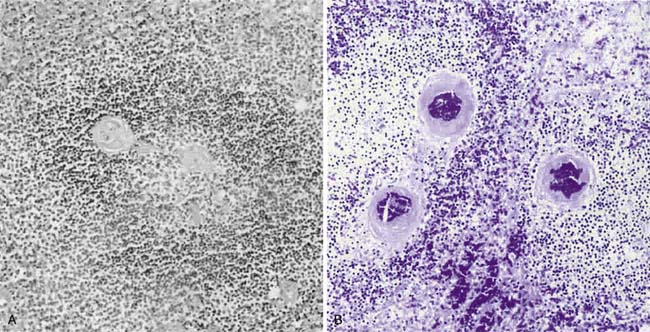
A prospective analysis by Hotchkiss et al.22 of 37 patients who died in a surgical intensive care unit and underwent immediate autopsy demonstrated increased lymphocytic and gut epithelial apoptosis in approximately 50% of septic patients with multiple organ dysfunction syndrome compared with essentially no alterations of cell death in critically ill, nonseptic patients (Figure 100-2). In addition, necrosis was detectable in 35% of patient livers but was minimal or absent in all other tissues examined. Similar findings have been shown in both neonates and pediatric patients with sepsis.23,24 Increased apoptosis in septic patients has been demonstrated to be associated with an increase in active caspase-3 activity. Further studies from this same group showed that B and CD4+ T lymphocytes are disproportionately lost in septic patients.25 B and CD4+ T-lymphocyte apoptosis is caspase-9 dependent, with the degree of cell death greater the longer the patient is septic. Antigen-presenting dendritic cells are also decreased in the spleens of septic patients.26 Apoptosis is significantly decreased, however, in macrophages obtained by bronchoalveolar lavage (BAL) in septic patients compared with nonseptic controls.27 This decrease in apoptosis is associated with lower levels of Bcl-2 than in control patients. An inverse correlation appears to exist between the severity of sepsis and the percentage of apoptotic alveolar macrophages. Minimal macrophage necrosis has been detected in BAL fluid.
Apoptosis is also decreased in neutrophils in patients who become septic after an initial traumatic insult, through upregulation of tyrosine phosphorylation by circulating mediators.28 This appears to be related to the septic insult because neutrophil apoptosis is not significantly different between noninfected trauma patients and healthy volunteers.28 In addition, circulating neutrophil apoptosis is decreased in infected patients with the systemic inflammatory response syndrome compared with controls. The decrease in neutrophil death is not associated with alterations in caspase-3 levels. Of note, however, levels of apoptosis are similar to those seen in patients who underwent elective abdominal aortic aneurysmectomy.29
Although there are few studies evaluating the role of autophagy in patients with sepsis, a recent study by Watanabe et al.30 showed a significant increase in autophagy in patients with sepsis. In this study, liver sections from six patients who died from sepsis were compared with four control patients. The patients who died from sepsis showed a threefold increase in autophagic vacuoles compared with controls.30 The significance of this finding is not entirely understood, and further studies are needed to determine the pathologic significance of increased autophagy in sepsis.
Noninfectious Inflammation
Similar to sepsis, tissue lymphocyte and intestinal epithelial apoptosis are increased in patients who have undergone shock and trauma. Intestinal specimens resected from 10 patients following motor vehicle collisions or gunshot wounds revealed extensive crypt epithelial and gut lymphocytic apoptosis, whereas control patients who underwent elective bowel resections had no obvious change in levels of cell death.31 Apoptosis was detectable within 2 hours of traumatic injury, and patients with the highest injury severity score had the most severe apoptosis. Importantly, patients with high apoptosis on initial evaluation following trauma had no evidence of increased cell death at follow-up elective laparotomy.
Increased apoptosis has also been demonstrated to be present in circulating T lymphocytes of patients following blunt trauma or burn injury.32,33 Although increased apoptosis of lymphocytes in the bloodstream was not directly associated with a negative outcome in the 30 individuals studied, patients with very high levels of apoptosis prior to complete activation and expansion of the T-cell response appear to be predisposed to anergy and organ failure. T-cell anergy leading to immunosuppression is further supported by Miller-Graziano et al.,34 who recently showed increased apoptosis in circulating T cells of trauma patients and that high levels of T-cell apoptosis correlated with T-cell anergy.
The apoptotic response of polymorphonuclear leukocytes to trauma has not been fully elucidated. Ogura et al.35 reported that apoptosis is decreased in this cell type for as long as 3 weeks following trauma. This contrasts with data showing that neutrophil apoptosis is decreased in septic trauma patients but is not significantly different between patients 24 hours after traumatic injury and control volunteers.28
Neutrophil apoptosis is low in BAL fluid from patients with acute respiratory distress syndrome (ARDS) as well as those at risk for ARDS.36,37 Levels of apoptosis did not correlate with patient survival. Interestingly, in vitro human polymorphonuclear leukocytes from healthy volunteers have less apoptosis when incubated with BAL fluid from ARDS patients compared with BAL fluid from healthy controls.
Further evaluation of this antiapoptotic effect on normal neutrophils showed it to be highest during the early stages of ARDS, with subsequent decreases over time. This change in apoptotic response to BAL fluid from ARDS patients correlates with levels of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor.38
Soluble Fas ligand is present in BAL fluid before and after onset of ARDS.39 However, its concentration is higher at the onset of ARDS in patients who eventually die. Of note, BAL fluid from patients with ARDS induces a Fas-dependent apoptosis in distal lung epithelial cells, whereas BAL fluid from patients at risk for ARDS but without the disease does not have an effect on distal lung epithelial cell death. BAL fluid also has elevated concentrations of the apoptosis-related molecules perforin, granzyme A, and granzyme B in critically ill patients with early ARDS compared with those not having lung injury or late ARDS.40
Although autophagy is thought to play a role in the human response to trauma and hemorrhage, there are currently no studies showing autophagy in humans. However, there is extensive literature on the role of autophagy in other noninfectious inflammatory conditions. Several groups have now shown a correlation between genetic mutation in autophagy genes and the development of inflammatory bowel disease.40,41 The exact mechanisms by which these specific mutations lead to inflammatory bowel disease are not known, but the role these autophagy genes play in inflammation is thought to be at least partially responsible.41 There is also growing evidence that autophagy is increased in atherosclerotic heart disease; however, it is unclear if this increase is detrimental or protective.43–45
Animal Studies
Sepsis
Similar to human autopsy studies, apoptosis is primarily localized to lymphocytes, dendritic cells, and the gut epithelium in animal models of sepsis. In cecal ligation and puncture (CLP), a murine model of ruptured appendicitis, as well as in overwhelming infection from Pseudomonas aeruginosa pneumonia, sampling of multiple cell and tissue types shows that maximal lymphocytic and intestinal apoptosis occurs 24 hours after onset of septic insult without substantial necrosis reported (Figure 100-3).46–51
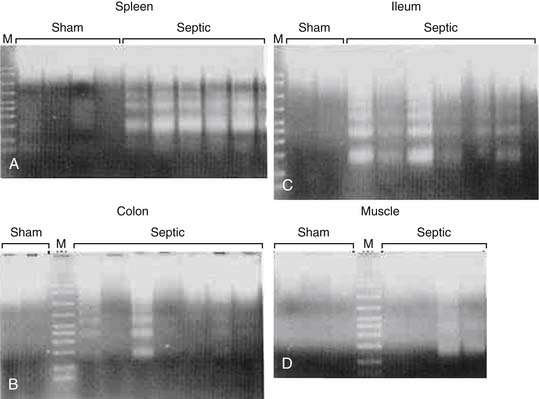
Figure 100–3 DNA agarose gel showing characteristic “laddering” pattern in spleen (A) and colon (B) in septic but not sham mice.
(From Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, et al: Apoptosis in lymphoid and parenchymal cells during sepsis: findings in normal and T- and B-cell-deficient mice, Crit Care Med 25:1298–1307, 1997.)





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


