109 Metabolic Acidosis and Alkalosis
 Acid-Base Disorders
Acid-Base Disorders
Normal Acid-Base Homeostasis
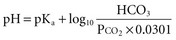
Under most circumstances, CO2 production and excretion are matched, and the usual steady-state PaCO2 is maintained at 40 mm Hg. Primary changes in PaCO2 can cause acidosis or alkalosis, depending on whether PaCO2 is above or below the normal value (respiratory acidosis or alkalosis, respectively). Underexcretion of CO2 produces hypercapnia, and overexcretion causes hypocapnia, both of which will affect the systemic pH. The PaCO2 is regulated primarily by neural respiratory factors and is not subject to regulation by the rate of CO2 production; therefore, hypercapnia is usually the result of hypoventilation rather than of increased CO2 production. Increases or decreases in PaCO2 may represent primary derangements of the ventilatory function of the lungs (under neural respiratory control) or may be due to compensatory changes in response to a primary alteration in the plasma [HCO3−].1
Primary alteration of PaCO2 evokes two metabolic mechanisms to limit change in systemic pH: the fast-acting cellular buffering and the renal-adaptive response, a slower process that becomes more efficient with time. This metabolic response would be secondary (or compensatory) to the primary respiratory disorder. A primary change in the plasma [HCO3−] as a result of metabolic or renal factors results in compensatory changes in ventilation that blunt the changes in blood pH that would occur otherwise. Such respiratory alterations are referred to as secondary or compensatory changes because they occur in response to primary metabolic alterations.1
The kidneys regulate plasma [HCO3−] through three main processes: (1) “reabsorption” of filtered HCO3−, (2) generation of “new” HCO3−, which is accomplished by formation of titratable acid, and (3) excretion of NH4+ in the urine. The kidney filters approximately 4000 mEq of HCO3− per day, and between 80% and 90% of HCO3− is reabsorbed in the proximal tubule. The distal nephron reabsorbs the remaining HCO3− and more importantly, secretes protons generated from dietary protein intake to defend systemic pH. Metabolism of the average diet rich in protein produces fixed acids that consume bicarbonate on entry into the extracellular fluid. Although the quantity of protons from dietary protein metabolism is small (40-60 mEq/day), it must be secreted to prevent chronic positive H+ balance and metabolic acidosis. This quantity of secreted protons (net acid) is represented in the urine as titratable acid and NH4+. Metabolic acidosis in the presence of normal renal function augments net acid excretion by markedly increasing NH4+ production and excretion. It is important to note that this vital compensatory mechanism is impaired in chronic renal failure, hyperkalemia, and renal tubular acidosis.1,2
 Diagnosis of Types of Disturbances
Diagnosis of Types of Disturbances
Historically, two different conceptual frameworks have evolved among clinicians and physiologists for interpreting acid-base phenomena. The traditional or bicarbonate-centered framework relies quantitatively on the Henderson-Hasselbalch equation (see later), whereas the Stewart or strong-ion approach utilizes the original Stewart equation to calculate the H+ concentration. The traditional approach has not only proven to be a mechanistic formulation that reflects the acid-base status at the tissue level but is also considerably easier to use in daily clinical practice, given the complexity of the Stewart theory and its associated fromulas.3
The most common clinical disturbances are simple acid-base disorders—that is, one of the metabolic disturbances (metabolic acidosis or alkalosis) or one of the respiratory disturbances (respiratory acidosis or alkalosis) occurring alone rather than in combination. Because physiologic compensation is not complete and cannot achieve a normal pH, the pH remains abnormal in simple disturbances. More complicated clinical situations can give rise to mixed acid-base disturbances through simultaneous expression of more than one simple disturbance, and in this setting the pH may be at a dangerous extreme or appear normal.1,3
Simple Acid-Base Disorders
The degree of primary alteration and secondary compensation in either or both of these two variables (acidosis or alkalosis) determines the systemic pH (acidemia or alkalemia). For example, metabolic acidosis due to an increase in endogenous acids (e.g., ketoacidosis) lowers extracellular fluid [HCO3−] and decreases systemic pH. This stimulates the medullary chemoreceptors to increase ventilation and to return the ratio of [HCO3−] to PaCO2, and thus pH, toward normal, although not to normal. Table 109-1 contains the acid-base disturbances along with the appropriate compensatory response for simple disorders. The degree of respiratory compensation expected in a simple form of metabolic acidosis can be predicted from the relationship PaCO2 = (1.5 × [HCO3−]) + 8 ± 2; that is, the PaCO2 is expected to decrease 1.25 mm Hg for each mEq/L per liter decrease in [HCO3−]. Thus, a patient with metabolic acidosis and [HCO3−] of 12 mEq/L would be expected to have a PaCO2 between 24 and 28 mm Hg. Values for PaCO2 below 24 or greater than 28 mm Hg define a mixed disturbance (metabolic acidosis plus respiratory alkalosis or metabolic acidosis plus respiratory acidosis, respectively). A readily available (though not as reliable) method of determining the nature and degree of compensatory response is the use of nomograms (Figure 109-1).1,4 If the arterial acid-base value falls within one of the shaded bands in Figure 109-1, one may assume that a simple acid-base disorder is present, and a tentative diagnostic category can be assigned. Values that fall outside the shaded area suggest the presence of a mixed disorder. These nomograms, though helpful, are not substitutes for an appreciation of the limits of compensation as displayed in Table 109-1.4

Mixed Acid-Base Disorders
Mixed acid-base disorders, defined as independently coexisting disorders, not merely compensatory responses, are more often seen in patients in ICUs and can lead to dangerous extremes of pH. A patient with diabetic ketoacidosis (DKA; high-AG metabolic acidosis) may develop an independent and superimposed respiratory problem leading to respiratory acidosis or alkalosis. Patients with underlying pulmonary disease may not respond to metabolic acidosis with an appropriate ventilatory response because of insufficient respiratory reserve. Such imposition of respiratory acidosis on metabolic acidosis can lead to severe acidemia and a poor outcome. When metabolic acidosis and metabolic alkalosis coexist in the same patient, the pH may be normal or near normal. When the pH is normal, an elevated AG (see later) denotes the presence of a metabolic acidosis. Patients who have ingested an overdose of drug combinations such as sedatives and salicylates may have mixed disturbances as a result of the acid-base response to the individual drugs (metabolic acidosis mixed with respiratory acidosis or respiratory alkalosis, respectively). Even more complex are triple acid-base disturbances. For example, patients with metabolic acidosis due to alcoholic ketoacidosis may develop metabolic alkalosis due to vomiting and superimposed respiratory alkalosis due to the hyperventilation of hepatic dysfunction or alcohol withdrawal.1 In general, a normal arterial pH in face of abnormal bicarbonate level, PaCO2, or AG is highly suggestive of a complex and mixed acid base disorder.
Pathway to Diagnosis of Acid-Base Disorders
A stepwise approach to the diagnosis of acid-base disorders follows and is summarized in Table 109-2. In the determination of arterial blood gases by the clinical laboratory, both pH and PaCO2 are measured, and the [HCO3−] is calculated from the Henderson-Hasselbalch equation. This calculated value should be compared with the measured [HCO3−] (or total CO2) on the electrolyte panel. These two values should agree within 2 mEq/L. If they do not, the values may not have been drawn simultaneously, a laboratory error may be present, or an error could have been made in calculating the [HCO3−]. After verifying the blood acid-base values, one can then identify the precise acid-base disorder.1
TABLE109-2 Steps in Acid-Base Diagnosis
2 Compare [HCO3−] on ABG and electrolytes to rule out error due to specimen handling or measurements. |
Adapted in part from DuBose TD Jr. Acid-base disorders. In: Brenner BM, editor Brenner and Rector’s the kidney 8th ed. Philadelphia: Saunders; 2008, p. 513.
Blood for electrolytes and arterial blood gases should be drawn simultaneously before therapy, because an increase in [HCO3−] occurs with metabolic alkalosis and respiratory acidosis. Conversely, a decrease in [HCO3−] occurs in metabolic acidosis and respiratory alkalosis.1,2
Metabolic acidosis often leads to hyperkalemia as a result of cellular shifts in which H+ is exchanged for K+ or Na+. For each decrease in blood pH of 0.10, the plasma [K+] should rise by 0.6 mEq/L. This relationship is not invariable, however. DKA, lactic acidosis, diarrhea, and renal tubular acidosis are regularly associated with potassium depletion because of urinary K+ wasting.1
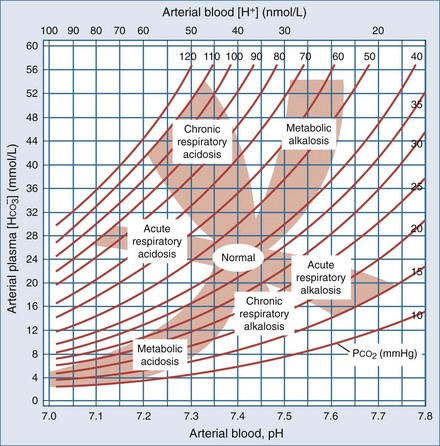
Anion Gap
Calculation of the anion gap is a key step in evaluation of acid-base disorders (see Table 109-2). Because normally the total unmeasured anions exceed the total unmeasured cations as indicated by the electrolyte panel, there exists an AG of 9 ± 3 mEq/L in plasma. The concentration of potassium in the blood usually is relatively small compared with that of sodium, chloride, and bicarbonate, so many clinicians omit this variable when calculating the AG.5
Simply put, AG represents unmeasured anions in plasma and is calculated as shown in the following equation. The various contributors to plasma AG in normal physiologic state and in metabolic acidosis are depicted in Figure 109-2. Patients with underlying pulmonary disease may not respond to metabolic acidosis with an appropriate ventilatory response because of insufficient respiratory reserve. Such imposition of respiratory acidosis on metabolic acidosis can lead to severe acidemia and a poor outcome. When metabolic acidosis and metabolic alkalosis coexist in the same patient, the pH may be normal or near normal. When the pH is normal, an elevated AG (see later) denotes the presence of a metabolic acidosis.
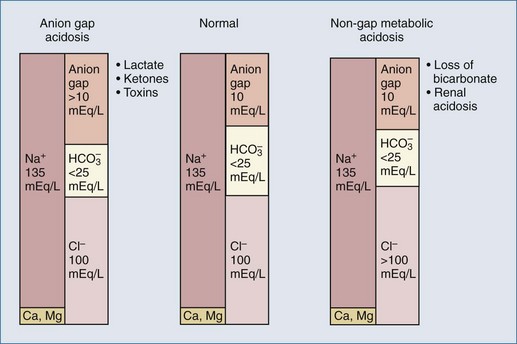
Figure 109-2 Contributors to plasma anion gap in normal physiologic state and in metabolic acidosis.
(Data from Gamble JL. Chemical anatomy, physiology, and pathology of extracellular fluid. 6th ed. Cambridge: Harvard University Press; 1954; and from Stewart PA. How to understand acid-base. New York: Elsevier; 1981.)
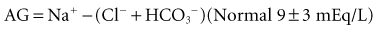
The unmeasured anions include predominately anionic proteins such as albumin but also phosphate, sulfate, and organic anions. An increase in the AG is most often due to an increase in unmeasured anions and less commonly is caused by a decrease in unmeasured cations (calcium, magnesium, potassium) (Table 109-3).1 When endogenously produced acid anions such as acetoacetate and lactate accumulate in extracellular fluid, the AG increases, causing a high-AG acidosis. In addition, the AG may increase with an increase in anionic albumin, either because of increased albumin concentration due to profound volume depletion or alkalosis, which alters albumin charge (increased negative charge).6
TABLE109-3 Anion Gap in the Diagnosis of Metabolic Acidosis Anion Gap = Na+ − (Cl− + HCO3−) = 9 + 3 mEq/L
| Decreased Anion Gap | Increased Anion Gap |
|---|---|
| Increased cations (not Na+): | Increased anions (not Cl− or HCO3−): |
| ↑ Ca++, Mg++ | ↑ Albumin concentration |
| ↑ Li+ | Alkalosis |
| ↑ IgG | ↑ Inorganic anions: |
| Decreased anions: | Phosphate |
| (not Cl− or HCO3−) | Sulfate |
| Hypoalbuminemia* | |
| Acidosis | ↑ Organic anions: |
| Laboratory error: | L-Lactate D-Lactate` |
| Hyperviscosity | Ketones |
| Bromism | Uremic |
| ↑ Exogenously supplied anions: | |
| Toxins: | |
| Salicylate | |
| Paraldehyde | |
| Ethylene glycol | |
| Methanol Toluene Pyroglutamic acid | |
| ↑ Unidentified anions: | |
| Uremic | |
| Hyperosmolar, nonketotic states | |
| Myoglobinuric acute renal failure | |
| Decreased cations (not Na+): | |
| ↓ Ca++, Mg++ |
* Albumin is the major unmeasured anion. A decline in serum albumin of 1.0 g/dL from the normal value of 4.5 g/dL decreases the anion gap by 2.3-2.5 mEq/L. Correction is very important to diagnose anion gap acidosis in setting of hypoalbuminemia.
Adapted from Emmett M, Narins RG. Clinical use of the anion gap. Medicine 1997;56:38-54; from Oh MS, Carroll HJ. The anion gap. N Engl J Med 1977;297:814-7; and from Kraut JA, Madisa NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol 2007;2:162-74. Epub 2006 Dec 6.
A low serum AG is not an uncommon occurrence and most frequently is the result of severe hypoalbuminemia. Albumin is the major contributor to serum AG, and a decline of 1 g/dL in serum albumin from the normal value of 4.5 g/dL will cause a reduction of 2.3 to 2.5 mEq/L in AG. Given the pivotal role of AG in formulating differential and treatment plans in acid-base disorders, it is extremely important to correct for low albumin when calculating AG in setting of hypoalbuminemia (see Table 109-3). Other causes of a low AG include:
Besides hypoalbuminemia, polyclonal gammopathy and monoclonal gammopathy with excessive accumulation of cationic immunoglobulin (Ig)G are the most common clinical disorders associated with a low serum AG. Therefore, once laboratory error and hypoalbuminemia have been excluded, a search for accumulation of IgG should be initiated. In patients with disturbed mentation or unexplained clinical findings, the possibility of lithium ingestion, bromism, or iodide intoxication should be considered. When the serum AG is negative in the absence of laboratory error, an extremely uncommon situation, bromide intoxication and iodide intoxication, should be excluded.5
In the face of a normal serum albumin, a high AG is usually due to non–chloride-containing acids that contain inorganic (phosphate, sulfate), organic (ketoacids, lactate, uremic organic anions), exogenous (salicylate or ingested toxins with organic acid production), or unidentified anions. As mentioned earlier, a high-AG acidosis has two identifying features: a low [HCO3−] and an elevated AG. The latter is present even if an additional acid-base disorder is superimposed to modify the [HCO3−] independently. Metabolic acidosis of the high-AG variety, concomitant with either chronic respiratory acidosis or metabolic alkalosis, represents a situation for which [HCO3−] may be normal or increased. Nevertheless, the AG is elevated, signaling the presence of the acidosis (see Table 109-3). In a typical simple AG metabolic acidosis, one would expect an equal but reciprocal change in serum bicarbonate and the AG, but this relationship does not hold when mixed disorders are present. Therefore, ΔAG versus ΔHCO3− is an important tool to search for a concealed acid-base disorder. For example, the combination of metabolic acidosis and metabolic alkalosis is expected to be present in a patient with advanced renal failure with several days’ history of vomiting. This mixed disorder would be most easily recognized when the AG is elevated but the HCO3− concentration and pH are near normal (ΔAG > ΔHCO3−). In general, a ΔAG/ΔHCO3− value of 1 is typical of pure high-AG acidosis such as lactic or DKA acidosis.7 A ratio significantly greater than 1 suggests the presence of metabolic acidosis and metabolic alkalosis, whereas a ratio less than 1 suggests the presence of mixed gap and non-gap metabolic acidosis. However, studies have indicated variability in the ΔAG/ΔHCO3−.5 This observation undercuts the ability to use this ratio alone to detect complex acid-base disorders, thus emphasizing the need to consider additional information to obtain the appropriate diagnosis.
 Metabolic Acidosis
Metabolic Acidosis
Metabolic acidosis can occur because of an increase in endogenous acid production (such as lactate and ketoacids), loss of bicarbonate (as in diarrhea), or accumulation of endogenous acids (as in renal failure). Metabolic acidosis along with an elevated AG have profound effects on patient survival.8
Effects of Acidosis
The effects of acidemia on the body are multiple (Table 109-4). The fall in blood pH is accompanied by a characteristic increase in ventilation, especially the tidal volume (Kussmaul respiration). Intrinsic cardiac contractility may be depressed, but inotropic function can be normal because of catecholamine release. Both peripheral arterial vasodilation and central venoconstriction can be present; the decrease in central and pulmonary vascular compliance predisposes to pulmonary edema with even minimal volume overload. CNS function is depressed, with headache, lethargy, stupor, and in some cases, even coma. Glucose intolerance may also occur.1
TABLE109-4 Systemic Effects of Acidosis
| Neurologic |
| Respiratory |
| Cardiovascular |
| Metabolic |
Adapted in part from Whitney GM, Szerlip HM. Acid-base disorder in critical care setting. In: DuBose TD, Hamm LL, editors. Acid-base and electrolytes disorders: a companion to Brenner and Rector’s the kidney. Philadelphia: Saunders; 2002, p. 165-83.
General Approach in Treatment of Metabolic Acidosis
The treatment of metabolic acidosis with alkali should be reserved for severe acidemia, except when the patient has no “potential [HCO3−]” in plasma. Potential [HCO3−] can be estimated from the increment (Δ) in the AG (ΔAG = patient’s AG − 10).1,9 It must be determined if the acid anion in plasma is metabolizable (i.e., β-hydroxybutyrate, acetoacetate, and lactate) or non-metabolizable (anions that accumulate in chronic renal failure and after toxin ingestion with subsequent kidney injury). The latter requires return of renal function to replenish the [HCO3−] deficit, a slow and often unpredictable process. Consequently, patients with a normal AG acidosis (hyperchloremic acidosis), a slightly elevated AG (mixed hyperchloremic and AG acidosis), or an AG attributable to a non-metabolizable anion in the presence of renal failure should receive alkali therapy, either orally (NaHCO3 or Shohl’s solution) or intravenously (NaHCO3), in an amount necessary to slowly increase the plasma [HCO3−] into the 20- to 22-mEq/L range. Controversy exists, however, in regard to the use of alkali in patients with a pure AG acidosis from accumulation of a metabolizable organic acid anion (ketoacidosis or lactic acidosis).1 In general, severe acidosis (pH < 7.15) warrants the intravenous (IV) administration of 50 to 100 mEq of NaHCO3 over 30 to 45 minutes during the initial 1 to 2 hours of therapy. Provision of such modest quantities of alkali in this situation seems to provide an added measure of safety, but it is essential to monitor plasma electrolytes during the course of therapy, because the [K+] may decline as pH rises. The goal is to increase the [HCO3−] to no more than 15 mEq/L and the pH to 7.25. The goal is never to increase these values to the normal values of 25 mEq/L and 7.40, respectively. It is important to point out that studies in humans and animals have failed to conclusively show any significant and positive effect with bicarbonate therapy on hemodynamic parameters or patient outcome in the ICU setting.1 However, the use of alkali remains a common practice in patients with profound acidosis or acidemia. There are two major clinical categories of metabolic acidosis: high AG and normal AG.
 High–Anion Gap Acidosis
High–Anion Gap Acidosis
High-AG acidosis is the most common form of metabolic acidosis encountered in the ICU. There are four principal causes of a high-AG acidosis (Figure 109-3; Tables 109-5 and 109-6)1,4:
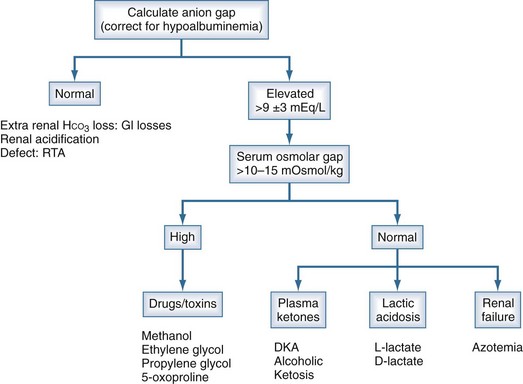
Figure 109-3 Anion gap acidosis workup.
(Data from Finkle KW, DuBose TD Jr. Metabolic acidosis. In: Dubose TD Jr, Hamm LL, editors. Acid-base and Electrolyte disorders: a companion to Brenner and Rector’s the kidney. Philadelphia: Saunders; 2002, p. 55-66.)
TABLE109-5 Clinical Causes of High Anion Gap and Normal Anion Gap Acidosis
| High Anion Gap |
| Normal Anion Gap |
Adapted in part from DuBose TD Jr. Acid-base disorders. In: Brenner BM, editor. Brenner and Rector’s the kidney. 8th ed. Philadelphia: Saunders; 2008, p. 513-46.
TABLE109-6 Etiologies of Lactic Acidosis
| L-Lactic Acidosis |
| D-Lactic Acidosis |
Adapted in part from DuBose TD Jr. Acid-base disorders. In: Brenner BM, editor. Brenner and Rector’s the kidney. 8th ed. Philadelphia: Saunders; 2008, p. 513-46.
Initial screening to identify the cause of the high-AG acidosis should include (1) a search in the history for evidence of drug or toxin ingestion (ethylene glycol, methyl alcohol, salicylates); (2) determination of whether diabetes mellitus is present (DKA); (3) a search for evidence of alcoholism or increased levels of β-hydroxybutyrate (alcoholic ketoacidosis); (4) observation for clinical signs of uremia and determination of the blood urea nitrogen and creatinine (uremic acidosis); (5) inspection of the urine for oxalate crystals (ethylene glycol); and (6) recognition of the common clinical settings in which lactate levels may be increased (hypotension, septic or hemorrhagic shock, cardiac failure, leukemia, cancer, and drug or toxin ingestion).1
Lactic Acidosis
Lactic acidosis is one the most common causes of high-AG acidosis in the ICU. An increase in plasma L-lactate is most commonly due to increased production of lactate in setting of an imbalance in oxygen supply and demand at the tissue level (type A). Thus type A lactic acidosis is thought to be caused by tissue hypoperfusion and/or severe hypoxemia, although in recent years this view is thought to be an oversimplification. Non-hypoxic conditions can also generate significant lactic acidosis (type B) in a variety of clinical settings such as malignancies, hepatic failure, or ingestion of drugs/toxins. The following are some of the causes of lactic acidosis in clinical setting (also see Table 109-6):
Among the most common causes of lactic acid acidosis in medical ICUs is unrecognized bowel ischemia or infarction in a patient with severe atherosclerosis or cardiac decomposition receiving vasopressors.10 Moreover, independent of the cause of hemodynamic instability, use of catecholamines, especially epinephrine, also results in lactic acidosis, presumably by stimulating cellular metabolism such as hepatic glycolysis.9
D-Lactic acid acidosis is due to formation of D-lactate by gut bacteria and may cause both an increased AG and hyperchloremia (see Table 109-6).9,10 This condition is caused by overgrowth of intestinal flora and may be associated with jejunoileal bypass or intestinal obstruction.
Lactic acidosis is among the most frequent and critical of all AG acidoses observed in the acute care setting. A study of 50 ICUs revealed an incidence of elevated lactate levels in over 60% of patients.11 Whether lactic acidosis represents a unique entity or is a consequence of a variety of other conditions common to the ICU has been debated. Lactate concentrations are mildly elevated in nonpathologic states (such as exercise), but the magnitude of elevation is generally small. For purposes of definition, a serum L-lactate level greater than 4 mEq/L (normal being 1 mEq/L) is thought to represent a clinically significant lactic acidosis and is the initiating point for resuscitative protocols in many critical care units. Nevertheless, some patients in the ICU maintain serum lactate levels between 2 to 4 mEq/L, and it is uncertain whether such patients progress to frank lactic acidosis, but studies are suggestive of higher mortality for patients with even intermediate rises in serum lactate.12
Whereas the lactic acid acidoses have been classified by Huckabee and Cohen into two types—type A (hypoxic) and type B (non-hypoxic) as noted earlier13—it has been recognized that lactic acidosis is often the result of the simultaneous existence of both hypoxic and non-hypoxic factors, and in many cases the precise etiology is difficult to establish. Decreased arterial perfusion to peripheral tissues in shock despite adequate arterial oxygen content, for example, results in L-lactic acid accumulation. Severe acidemia decreases portal blood flow and hepatic clearance of lactic acid.1 Moreover, in sepsis there is both a decrease in tissue perfusion and a decrease in oxygen utilization. Technically, therefore, the classification of lactic acidosis is primarily of conceptual interest.
Numerous drugs have been implicated in the occurrence of lactic acidosis (see Table 109-6). Of particular note is biguanide (metformin) and antiretroviral therapy, specifically the nucleoside reverse transcriptase inhibitors (NRTIs) used in treatment of HIV infection. The biguanide family of hypoglycemic agents, which includes metformin, have been associated with mild elevation in serum lactate (usually <2 mEq/L) in patients with otherwise normal renal and hepatic function. Cases of severe lactic acidosis associated with use of metformin are typically accompanied by presence of sepsis and/or profound renal failure. The mechanism is largely unknown,14 and the current recommendations are that the drug not be used in patients with congestive heart failure, liver disease, and significant renal insufficiency (creatinine >1.5 mg/dL in men, or >1.4 mg/dL in women).9,15 Nucleoside analogs used in treatment of HIV infections inhibit mitochondrial polymerase and lead to lactic acid accumulation. Hyperlactemia is common with NRTI therapy, especially stavudine and zidovudine, but the serum lactate is mildly elevated and well compensated. Risk factors for NRTI therapy–associated lactic acidosis include a creatinine clearance less than 70 mL/min and a low CD4+ T-lymphocyte count.9,16
Carbon monoxide poisoning produces lactic acidosis by reducing the oxygen-carrying capacity of the hemoglobin, resulting in tissue hypoxia. A newly recognized and not too uncommon cause of lactate accumulation is propylene glycol. This agent is used as carrier for a variety of IV medications used in ICU setting, most notably diazepam, lorazepam, nitroglycerin, and etomidate. Metabolism of propylene glycol by alcohol dehydrogenase in the liver results in lactate formation, which is then converted to pyruvate and shunted to glycolytic pathways. There have been numerous reports of high-AG acidosis and elevated serum osmolarity in patients receiving benzodiazepine infusions in the critical care setting.17
Critically ill patients with a significantly elevated AG or low serum bicarbonate should be suspected of having a lactic acidosis, particularly in the presence of hepatic insufficiency. A high index of suspicion must be maintained, however, because the AG is a relatively insensitive reflection of lactic acidosis. Iberti and coworkers reported a poor correlation between arterial pH, the AG, and serum lactate levels.18 Fifty percent of patients with serum lactate levels above 5 and less than 9.9 mmol/L displayed a normal AG.
Several investigators have sought to characterize the prognostic value of serum lactic acid levels. Studies have found an inverse correlation between mortality and L-lactate levels above 2.0 to 2.5 mmol/L.19–21 Prognosis is related to lactate concentration, as well as the ability to metabolize a lactic acid load after a resuscitative effort. Although lactic acidemia is associated with adverse outcome during critical illness, it does not appear to be a direct causative agent, rather a marker of poor prognosis. Therapeutic interventions that target the primary pathophysiology rather than the lactic acidosis per se have been shown to have outcome benefits.22 Moreover, normalization of elevated lactic acid levels regardless of causative source is associated with better outcome in patients with sepsis, but this may be a surrogate for the acuity of the illness and index of organ dysfunction.23 Falk and associates observed that the ability to lower serum lactate levels by 50% within 18 hours after resuscitation correlated with a significantly greater rate of survival.24 Other studies have reinforced these findings, revealing both significantly lower lactate levels and increased ability to clear lactate in survivors as opposed to nonsurvivors.25 To add to this argument, dichloroacetate, which indirectly decreases lactic acid level, has not been shown to improve survival, suggesting that elevated lactate level is an epiphenomenon with varying prognostic value in different clinical settings.26
Treatment of Lactic Acidosis
Therapy of lactic acidosis has two distinct goals. The first is to identify and remedy the defect in the oxidative metabolism (hypoperfusion and hypoxemia being the most common causes) in order to halt further production of lactate. The second is to raise the serum pH toward normal. If the underlying pathophysiologic state is effectively treated, the excess lactate production and acidemia often correct without specific interventions. As mentioned earlier, there is controversy as to whether lactic acidosis directly contributes to mortality or is simply a marker of the severity of the underlying illness,9,10 and this has led to added debate on recommendations for use of buffers in the management of lactic acidosis. Nevertheless, the basic principle and most effective therapy for L-lactic acidosis is that the underlying condition disrupting the normal lactate metabolism must first be corrected.
Fluid overload occurs rapidly with NaHCO3 administration because of the massive amounts required in some cases, along with high sodium content of the solution. In addition, central venoconstriction and decreased cardiac output are common, which lead to tissue hypoperfusion and further end-organ dysfunction—specifically, reduced glomerular filtration. As such, the volume overload and acidemia may precipitate the need for renal replacement therapy which can simultaneously deliver HCO3−, remove lactate and excess extracellular fluid (ECF) volume, and correct electrolyte abnormalities. Although use of continuous hemofiltration (HF) in critically ill patients provides minimal additional lactate clearance, perhaps due to the high rate of its production,27 the ultrafiltration accomplished with continuous renal replacement therapy may offer additional benefits in terms of volume and electrolyte management. The use of bicarbonate-based replacement fluid or dialysate is thought to convey better acid-base control when compared to lactate based solutions, though controlled studies have shown controversial results.28,29 There are theoretical advantages of extracorporeal therapies such as HF or hemodiafiltration in treatment of sepsis with lactic acidosis. These include enhanced removal of inflammatory mediators and endotoxin while providing early and effective management of volume and acid-base disturbances. However, as yet, the evidence in humans is too limited to recommend HF as an adjunctive therapy for critically ill patients with sepsis or systemic inflammatory response syndrome (SIRS). Regarding the many uncertainties about optimal volume (high or very high) and type of membrane, clinical studies should first focus on endpoints as recovery from organ failure and length of treatment before survival studies are started.30 Recently, smaller uncontrolled trials have shown promising effects of HF in refractory shock,31 which raises the need for additional large-scale studies to further define the role of renal replacement therapy in treatment of sepsis and shock.
If the underlying cause of the L-lactic acidosis can be remedied, it is anticipated that the lactate will be reconverted to HCO3−. HCO3− derived from lactate conversion, and any new HCO3− generated by renal mechanisms during acidosis and IV-administered bicarbonate are all additive and may result in overshoot alkalosis.32
Sodium Bicarbonate
Despite highly debated and somewhat discouraged practice of its use, bicarbonate therapy remains the most commonly administered agent in treatment of lactic acidosis. Two major factors contribute to this common practice: bicarbonate is readily available to physicians who feel obliged to react to a low pH; and bicarbonate use is often associated with some degree of rather immediate improvement in measured pH which may, often unrealistically, be interpreted as a sign of improvement in the metabolic derangements and patient condition. Although severe acidosis is classically thought to have a deleterious effect on cardiopulmonary performance, recent studies have actually shown an improvement in cardiac performance in the presence of a mild to moderate acidosis.10 Accordingly, the pH below which most clinicians feel obligated to use NaHCO3 has declined. Therefore, the recommendation for administration of NaHCO3 in the treatment of severe acidosis when the pH is less than 7.15 seems reasonable. At this pH, as predicted by the Henderson-Hasselbalch equation, minor changes in bicarbonate or PCO2 will result in a large decrease in pH.33 However, there are no data supporting a specific pH at which therapy must be instituted.
A prospective study evaluating NaHCO3 in patients with lactic acidosis showed an increase in serum pH but no improvement in hemodynamics when compared with normal saline.34 The use of NaHCO3 also failed to increase hemodynamic responsiveness to circulating catecholamines concomitant with a decrease in serum ionized calcium.35–38 It is important to note, however, the numerous deleterious effects of bicarbonate therapy in treatment of lactic acidosis. NaHCO3 therapy can cause fluid overload and hypertension, because the amount required can be massive when accumulation of lactic acid is relentless. Fluid administration is poorly tolerated because of central venoconstriction, especially in the oliguric patient, and can worsen the state of volume overload. If the underlying cause of the lactic acidosis can be remedied, blood lactate will be converted to HCO3− and may result in an overshoot alkalosis. Sodium bicarbonate may also result in impaired utilization of oxygen and increase anaerobic metabolism through stimulation of phosphofructokinase (by raising cellular pH), which leads to further lactate accumulation. Finally, sodium bicarbonate IV administration generates CO2 (HCO3− + H+ → H2O + CO2). With depressed cardiac output or ventilatory capacity of lungs, CO2 can accumulate, causing intracellular acidosis and a further reduction in cardiac output. Other possible adverse effects of bicarbonate infusion include hypernatremia, hyperosmolarity, hypocalcaemia, and hypokalemia.
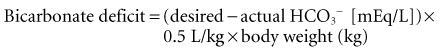
This must be viewed as a very rough estimate with considerable variability in the clinical setting. Constant infusion of hypertonic bicarbonate has many disadvantages and is discouraged to minimize the rapid development of fluid overload. The use of continuous renal replacement therapy is common in patients with overt shock and lactic acidosis, as frequently simultaneous acute kidney injury exists in such a setting. Though the extracorporeal removal of lactate is negligible and likely bears no effect on treatment of underlying cause,27 effective alkali therapy can be provided with minimal volume overload using the newer bicarbonate-based dialysate or replacement fluid formulations.


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


