Mechanisms of Anesthesia and Consciousness
C. Michael Crowder
Ben Julian Palanca
Alex S. Evers
Key Points
Related Matter
Meyer-Overton Rule
The introduction of general anesthetics into clinical practice over 150 years ago stands as one of the seminal innovations of medicine. This single discovery facilitated the development of modern surgery and spawned the specialty of anesthesiology. Despite the importance of general anesthetics and despite more than 100 years of active research, the molecular mechanisms responsible for anesthetic action remain one of the unsolved mysteries of science.
Why have mechanisms of anesthesia been so difficult to elucidate? Anesthetics, as a class of drugs, are challenging to study for three major reasons:
Anesthesia, by definition, is a change in the responses of an intact animal to external stimuli. Making a definitive link between anesthetic effects observed in vitro and the anesthetic state observed and defined in vivo has proven difficult.
No structure–activity relationships are apparent among anesthetics; a wide variety of structurally unrelated compounds, ranging from steroids to elemental xenon, are capable of producing clinical anesthesia. This suggests that there are multiple molecular mechanisms that can produce clinical anesthesia.
Anesthetics work at very high concentrations in comparison to drugs, neurotransmitters, and hormones that act at specific receptors. This implies that if anesthetics do act by binding to specific receptor sites, they must bind with very low affinity and probably stay bound to the receptor for very short periods of time. Low-affinity binding is much more difficult to observe and characterize than high-affinity binding.
Despite these difficulties, molecular and genetic tools are now available that should allow for major insights into anesthetic mechanisms in the next decade. The aim of this chapter is to provide a conceptual framework for the reader to catalog current knowledge and integrate future developments about mechanisms of anesthesia. Five specific questions will be addressed in this chapter:
What is anesthesia and how do we measure it?
What are the molecular targets of anesthetics?
What are the cellular neurophysiologic mechanisms of anesthesia (e.g., effects on synaptic function vs. effects on action potential generation) and what anesthetic effects on ion channels and other neuronal proteins underlie these mechanisms?
How are the molecular and cellular effects of anesthetics linked to the behavioral effects of anesthetics observed in vivo?
What are the major anatomic sites of anesthetic action in the CNS and how do anesthetics interfere with their interactions?
What Is Anesthesia?
Regardless of which definition of anesthesia is used, rapid and reversible drug-induced changes in behavior or perception are essential to anesthesia. As such, anesthesia can only be defined and measured in the intact organism. Changes in behavior such as unconsciousness or amnesia can be intuitively understood in higher organisms such as mammals, but become increasingly difficult to define as one descends the phylogenetic tree. Thus, while anesthetics have effects on organisms ranging from worms to man, it is difficult to map with certainty the effects of anesthetics observed in lower organisms to any of our behavioral definitions of anesthesia. This contributes to the difficulty of using simple organisms as models in which to study the molecular mechanisms of anesthesia. Similarly, any cellular or molecular effects of anesthetics observed in higher organisms can be extremely difficult to link with the constellation of behaviors that constitute the anesthetic state. The absence of a simple and concise definition of anesthesia
has clearly been one of the stumbling blocks to elucidating the mechanisms of anesthesia at a molecular and cellular level. Precise definitions for each of the component behaviors of the anesthetic state will be an important tool in dissecting the molecular and cellular mechanisms of each of the clinically important effects of anesthetic agents.
has clearly been one of the stumbling blocks to elucidating the mechanisms of anesthesia at a molecular and cellular level. Precise definitions for each of the component behaviors of the anesthetic state will be an important tool in dissecting the molecular and cellular mechanisms of each of the clinically important effects of anesthetic agents.
An additional difficulty in defining anesthesia is that our understanding of the mechanisms of consciousness is rather amorphous at present. One cannot easily define anesthesia when the neurobiologic phenomena ablated by anesthesia are not well understood. As discussed later in this chapter, the neural substrates for consciousness are beginning to be unraveled1 and new theories2 have incorporated this new anatomic knowledge leading to identification of surrogate physiologic markers of consciousness.3 These new insights into mechanisms of consciousness are discussed in the section Where in the Central Nervous System Do Anesthetics Work?
Finally, it has long been assumed that anesthesia is a state that is achieved when an anesthetic agent reaches a specific concentration at its effect site in brain and that if tolerance to the anesthetic develops, increasing concentrations of anesthetic might be required to maintain a constant level of anesthesia during prolonged anesthetic administration. The recent finding that it takes a higher anesthetic brain concentration to induce anesthesia than to maintain anesthesia (i.e., emergence occurs at a significantly lower concentration than induction) contradicts these assumptions.4 This phenomenon, referred to as neural inertia, adds a wrinkle to the definition of anesthesia and suggests that the mechanisms of anesthetic induction and emergence may be different.
How Is Anesthesia Measured?
Because of the limitations of MAC, monitors that measure some correlate of anesthetic depth have been introduced into clinical practice.8 The most popular of these monitors converts spontaneous electroencephalogram (EEG) waveforms into a single value that correlates with anesthetic depth for some general anesthetics. To date, these monitors have not been shown to be more effective at preventing awareness during anesthesia than simply maintaining an adequate end-tidal anesthetic concentration9,10 or giving a standard dose of intravenous anesthetic. Nonetheless it is logical to think that different individuals may have different sensitivities to anesthetics and that measuring a surrogate end point such as a processed EEG value,11 an evoked potential,12 or a functional neuroimaging signal indicative of integrated cortical activity1 might be a better indicator of anesthetic depth than merely measuring delivered concentration.
What is the Chemical Nature of the Anesthetic Target Sites?
The Meyer–Overton Rule
Exceptions to the Meyer–Overton Rule
Compounds exist that are structurally similar to halogenated anesthetics,16 barbiturates,17 and neurosteroids,18 yet are convulsants rather than anesthetics. On the basis of olive oil/gas partition coefficients of the halogenated convulsant compounds,
anesthesia should have been achieved within the range of concentrations studied.19 Halogenated compounds have also been identified that are neither anesthetic nor convulsant despite oil/gas partition coefficients that would predict they should be anesthetics.19 Interestingly, some of these polyhalogenated compounds do produce amnesia in animals20 and are thus referred to as non-immobilizers rather than as non-anesthetics. Finally, polyhalogenated alkanes, referred to as transitional compounds, have been identified that anesthetize mice, but only at concentrations 10 times those predicted by their oil/gas partition coefficients.19
anesthesia should have been achieved within the range of concentrations studied.19 Halogenated compounds have also been identified that are neither anesthetic nor convulsant despite oil/gas partition coefficients that would predict they should be anesthetics.19 Interestingly, some of these polyhalogenated compounds do produce amnesia in animals20 and are thus referred to as non-immobilizers rather than as non-anesthetics. Finally, polyhalogenated alkanes, referred to as transitional compounds, have been identified that anesthetize mice, but only at concentrations 10 times those predicted by their oil/gas partition coefficients.19
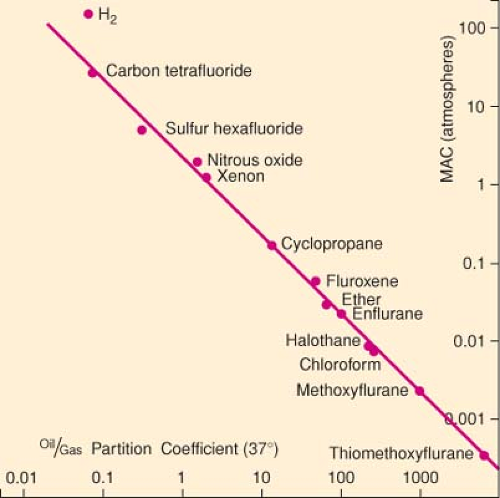 Figure 5.1. The Meyer–Overton rule. There is a linear relationship (on a log–log scale) between the oil/gas partition coefficient and the anesthetic potency (MAC) of a number of gases. The correlation between lipid solubility and MAC extends over a 70,000-fold difference in anesthetic potency. (Reproduced with permission from Tanfiuji Y, Eger EI, Terrell RC. Some characteristics of an exceptionally potent inhaled anesthetic: thiomethoxyflurane. Anesth Analg. 1977;56:387.) |
In several homologous series of anesthetics, anesthetic potency increases with increasing chain length until a certain critical chain length is reached. Beyond this critical chain length, compounds are unable to produce anesthesia, even at the highest attainable concentrations. In the series of n-alkanols, for example, anesthetic potency increases from methanol through dodecanol; all longer alkanols are unable to produce anesthesia.21 This phenomenon is referred to as the cutoff effect. Cutoff effects have been described for several homologous series of anesthetics including n-alkanes, n-alkanols, cycloalkanemethanols,22 and perfluoroalkanes.23 While the anesthetic potency in each of these homologous series of anesthetics shows a cutoff, a corresponding cutoff in octanol/water or oil/gas partition coefficients has not been demonstrated. Therefore, compounds above the cutoff represent a deviation from the Meyer–Overton rule.
A final deviation from the Meyer–Overton rule is the observation that enantiomers of anesthetics differ in their potency as anesthetics. Enantiomers (mirror-image compounds) are a class of stereoisomers that have identical physical properties, including identical solubility in solvents such as octanol or olive oil. Animal studies of barbiturate anesthetics,24,25 ketamine,26 neurosteroids,27 etomidate,28 and isoflurane29 all show enantioselective differences in anesthetic potency. These differences in potency range in magnitude from a more than 10-fold difference between the enantiomers of etomidate or the neurosteroids to a 60% difference between the enantiomers of isoflurane. It is argued that a major difference in anesthetic potency between a pair of enantiomers can only be explained by a protein-binding site (see Protein Theories of Anesthesia); this appears to be the case for etomidate and the neurosteroids.
The exceptions to the Meyer–Overton rule do not obviate the importance of the rule. They do, however, indicate that the properties of a solvent such as octanol describe some, but not all, of the properties of an anesthetic-binding site. Compounds that deviate from the Meyer–Overton rule suggest that anesthetic target site(s) are also defined by other properties including size and shape.
In defining the molecular target(s) of anesthetic molecules, one must be able to account both for the Meyer–Overton rule and for the well-defined exceptions to this rule. It has sometimes been suggested that a correct molecular mechanism of anesthesia should also be able to account for pressure reversal. Pressure reversal is a phenomenon whereby the concentration of a given anesthetic needed to produce anesthesia is greatly increased if the anesthetic is administered to an animal under hyperbaric conditions. The idea that pressure reversal is a useful tool for elucidating mechanisms of anesthesia is based on the assumption that pressure reverses the specific physicochemical actions of the anesthetic that are responsible for producing anesthesia; that is to say, pressure and anesthetics act on the same molecular targets. However, evidence suggests that pressure reverses anesthesia by producing excitation that physiologically counteracts anesthetic depression, rather than by acting as an anesthetic antagonist at the anesthetic site of action.30 Therefore, in the ensuing discussion of molecular targets of anesthesia, pressure reversal will not be further mentioned.
Lipid versus Protein Targets
Anesthetics might interact with several possible molecular targets to produce their effects on the function of ion channels and other proteins. Anesthetics might dissolve in the lipid bilayer, causing physicochemical changes in membrane structure that alter the ability of embedded membrane proteins to undergo conformational changes important for their function. Alternatively, anesthetics could bind directly to proteins (either ion channel proteins or modulatory proteins), thus either1 interfering with binding of a ligand (e.g., a neurotransmitter, a substrate, a second messenger molecule) or2 altering the ability of the protein to undergo conformational changes important for its function. The following section summarizes the arguments for and against lipid theories and protein theories of anesthesia.
Lipid Theories of Anesthesia
In its simplest incarnation, the lipid theory of anesthesia postulates that anesthetics dissolve in the lipid bilayers of biologic membranes and produce anesthesia when they reach a critical concentration in the membrane. Consistent with this hypothesis, the membrane/gas partition coefficients of anesthetic gases in pure lipid bilayers correlate strongly with anesthetic potency.31 Also, consistent with the lipid theories, various membrane perturbations are produced by general anesthetics; however, the magnitude of these changes produced by clinical concentrations of anesthetics are quite small and are thought to be very unlikely to disrupt nervous system
function.32 While some of the more sophisticated lipid theories can account for the cutoff effect and for the ineffectiveness of non-immobilizers, no lipid theory can plausibly explain all anesthetic pharmacology. Thus, most investigators do not consider lipids as the most likely target of general anesthetics.
function.32 While some of the more sophisticated lipid theories can account for the cutoff effect and for the ineffectiveness of non-immobilizers, no lipid theory can plausibly explain all anesthetic pharmacology. Thus, most investigators do not consider lipids as the most likely target of general anesthetics.
Protein Theories of Anesthesia
The Meyer–Overton rule could also be explained by the direct interaction of anesthetics with hydrophobic sites on proteins. Three types of hydrophobic sites on proteins might interact with anesthetics:
Hydrophobic amino acids comprise the core of water-soluble proteins. Anesthetics could bind in hydrophobic pockets that are fortuitously present in the protein core.
Hydrophobic amino acids also form the lining of binding sites for hydrophobic ligands. For example, there are hydrophobic pockets in which fatty acids tightly bind on proteins such as albumin and the low-molecular-weight fatty acid–binding proteins. Anesthetics could compete with endogenous ligands for binding to such sites on either water-soluble or membrane proteins.
Hydrophobic amino acids are major constituents of the α-helices, which form the membrane-spanning regions of membrane proteins; hydrophobic amino acid side chains form the protein surface that faces the membrane lipid. Anesthetic molecules could interact with pockets formed between the α-helices or with the hydrophobic surface of these membrane proteins, disrupting normal lipid–protein interactions and possibly directly affecting protein conformation.
Evidence for Anesthetic Binding to Proteins
A breakthrough in protein theories of anesthesia was the demonstration that a purified water-soluble protein, firefly luciferase, could be inhibited by general anesthetics. This provided the important proof of principle that anesthetics could bind to proteins in the absence of membranes. Numerous studies have extensively characterized the anesthetic inhibition of firefly luciferase activity and have shown that inhibition occurs at concentrations very similar to those required to produce clinical anesthesia, is consistent with the Meyer–Overton rule, is competitive with respect to the substrate D-luciferin, and exhibits a cutoff in anesthetic potency for both n-alkanes and n-alkanols.33,34 These data suggest that the luciferin-binding pocket may have physical and chemical characteristics similar to those of a putative anesthetic-binding site in the CNS. To address proteins more relevant to anesthetic effects on the nervous system, numerous studies have employed site-directed mutagenesis of anesthetic-sensitive ion channels to identify amino acid residues that are crucial to anesthetic action. While the residues identified in these studies may contribute to anesthetic-binding sites, they may alternatively be sites that are essential for anesthetic-induced conformational changes in the protein. The literature on site-directed mutagenesis studies to identify putative anesthetic-binding sites on ion channels is extensively reviewed in the section Anesthetic Actions on Ion Channels.
More direct approaches to study anesthetic binding to proteins have included NMR spectroscopy and photoaffinity labeling. Early studies using 19F-NMR spectroscopy demonstrated that isoflurane binds to the fatty acid–binding sites on bovine serum albumin (BSA) and that binding is competitively inhibited by halothane, methoxyflurane, sevoflurane, and octanol.35,36 Using this BSA model, it was subsequently shown that anesthetic-binding sites could be identified and characterized using photoaffinity labeling with 14C-labeled halothane.37,38 Photoaffinity-labeling reagents have subsequently been developed for a variety of anesthetics including etomidate, propofol,39,40 and neurosteroids and are a useful tool to validate results obtained using site-directed mutagenesis as well as to identify novel binding sites.
The most extensive photolabeling studies have used etomidate analogue photolabeling reagents to identify etomidate-binding sites on purified GABAA receptors. An initial study used azietomidate, a photolabeling reagent that preferentially labels nucleophilic amino acids, to photolabel-purified GABAA receptors from bovine brain.41 This study identified two methionine residues that were sites of attachment for azi-etomidate: One site on the TM1 helix (Met-236) of the α1 subunit and the other (Met-286) on the TM3 helix of the β3 subunit. These data suggest an etomidate-binding pocket in the transmembrane domain at the interface between the α1 and β3 subunits. A subsequent study using TDBzl-etomidate, a photolabeling reagent with broader amino acid side chain reactivity, identified additional amino acids that confirmed and further defined this inter-subunit–binding site.42 The combined results of site-directed mutagenesis studies and photoaffinity-labeling studies identified a specific, functionally relevant binding site for etomidate on GABAA receptors, definitively refuting lipid theories of anesthetic action. Photoaffinity-labeling studies with other anesthetic agents should provide similar levels of detail regarding anesthetic-binding sites on protein targets.
Although NMR and photoaffinity techniques can provide extensive information about anesthetic-binding sites on proteins, they cannot reveal the details of the three-dimensional structure of these sites. X-ray diffraction crystallography can provide this kind of three-dimensional detail and has been used to study anesthetic interactions with a small number of proteins. Firefly luciferase has been crystallized in the presence and absence of the anesthetic bromoform, confirming that anesthetics bind in the D-luciferin–binding pocket.43 The firefly luciferase data demonstrate that anesthetics can bind to endogenous ligand–binding sites and that this binding strongly correlates with anesthetic inhibition of protein function. Human serum albumin has also been crystallized in the presence of either propofol or halothane, demonstrating binding of both anesthetics to preformed fatty acid–binding pockets. While these data provide insight into
the structure of anesthetic-binding sites, x-ray crystallographic studies of anesthetic-binding sites on biologically relevant targets such as ion channels have been hampered by difficulties with crystallizing membrane proteins. Recently, a bacterial homolog of the ligand-gated ion channels, GLIC, has been crystallized and its crystal structure has been solved.44 GLIC has been shown to be sensitive to clinical concentrations of anesthetics and the crystal structure of GLIC complexed with either desflurane or propofol has been solved.45 These data reveal a preformed binding cavity in the interface between the transmembrane domains of each subunit of the ion channel. This landmark study opens the way to define the precise dimensions and location of anesthetic-binding pockets on their target proteins.
the structure of anesthetic-binding sites, x-ray crystallographic studies of anesthetic-binding sites on biologically relevant targets such as ion channels have been hampered by difficulties with crystallizing membrane proteins. Recently, a bacterial homolog of the ligand-gated ion channels, GLIC, has been crystallized and its crystal structure has been solved.44 GLIC has been shown to be sensitive to clinical concentrations of anesthetics and the crystal structure of GLIC complexed with either desflurane or propofol has been solved.45 These data reveal a preformed binding cavity in the interface between the transmembrane domains of each subunit of the ion channel. This landmark study opens the way to define the precise dimensions and location of anesthetic-binding pockets on their target proteins.
Summary
What is the stoichiometry of anesthetic binding to a protein (i.e., do many anesthetic molecules interact with a single protein molecule or only a few)?
Do anesthetics compete with endogenous ligands for binding to hydrophobic pockets on protein targets or do they bind to fortuitous cavities in the protein?
Do all anesthetics bind to the same pocket on a protein or are there multiple hydrophobic pockets for different anesthetics?
How many proteins have hydrophobic pockets in which anesthetics can bind at clinically used concentrations?
How Do Anesthetics Interfere with the Electrophysiologic Function of the Nervous System?
The functional unit of the CNS is the neuron and ultimately general anesthetics must disrupt the function of neurons’ mediating behavior, consciousness, and memory. In the simplest terms, anesthetics could accomplish this by altering the intrinsic firing rate of individual neurons, termed neuronal excitability, and/or by altering communication between neurons, generally occurring via synaptic transmission.
Neuronal Excitability
Synaptic Transmission
a great deal of heterogeneity in the manner in which anesthetic agents affect different synapses. This is not surprising given the large variation in synaptic structure, function (i.e., efficacy), and chemistry (neurotransmitters, modulators) extant in the nervous system.
Presynaptic Effects
Neurotransmitter release from glutamatergic synapses has consistently been found to be inhibited by clinical concentrations of volatile anesthetics. For example, a study by Perouansky and colleagues66 conducted in mouse hippocampal slices showed that halothane inhibited excitatory postsynaptic potentials elicited by presynaptic electrical stimulation, but not those elicited by direct application of glutamate. This indicates that halothane must be acting to prevent the release of glutamate. MacIver and colleagues extended these observations by finding that the inhibition of glutamate release from hippocampal neurons is not due to effects at GABAergic synapses that could indirectly decrease transmitter release from glutamatergic neurons. Reduction of glutamate release by intravenous anesthetics has also been demonstrated, but the evidence is more limited and the effects potentially indirect.67,68
The data for anesthetic effects on inhibitory neurotransmitter release are mixed. Inhibition,69 stimulation,70,71 and no effect72 on GABA release have been reported for both volatile and intravenous anesthetics. In a brain synaptosomal preparation where both GABA and glutamate release could be studied simultaneously, Westphalen and Hemmings73 found that glutamate and, to a lesser degree, GABA release were inhibited by clinical concentrations of isoflurane. The mechanism underlying the anesthetic effects on transmitter release has not been established. The mechanism does not appear to involve reduced neurotransmitter synthesis or storage, but rather is a direct effect on neurosecretion. A variety of evidence argues that at some synapses a substantial portion of the anesthetic effect is upstream of the transmitter release machinery, perhaps on presynaptic sodium channels or potassium leak channels (see later discussion). However, genetic data in Caenorhabditis elegans show that mutations in the transmitter release machinery strongly influence volatile anesthetic sensitivity.74,75 Recent evidence where the homologous rat mutant protein also reduced volatile anesthetic sensitivity against transmitter release suggests that this mechanism may be conserved in mammals.76
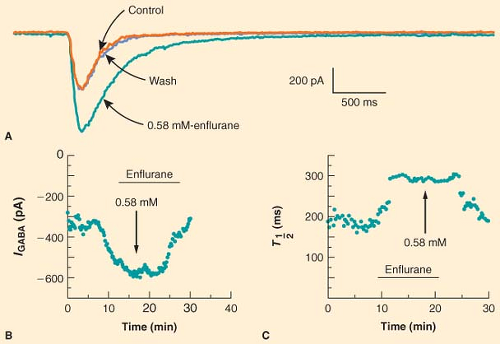 Figure 5.2. Enflurane potentiates the ability of GABA to activate a chloride current in cultured rat hippocampal cells. This potentiation is rapidly reversed by removal of enflurane (wash; A). Enflurane increases both the amplitude of the current (B) and the time (T1/2) it takes for the current to decay (C). (Reproduced with permission from Jones MV, Brooks PA, Harrison L: Enhancement of GABA-activated Cl− currents in cultured rat hippocampal neurons by three volatile anaesthetics. J Physiol. 1992;449:289.) |
Postsynaptic Effects
Anesthetics alter the postsynaptic response to released neurotransmitter at a variety of synapses. Anesthetic modulation of excitatory neurotransmitter receptor function varies depending on the receptor type, anesthetic agent, and preparation. In a classic study, Richards and Smaje77 examined the effects of several anesthetic agents on the response of olfactory cortical neurons to application of glutamate, the major excitatory neurotransmitter in the CNS. They found that while pentobarbital, diethyl ether, methoxyflurane, and alphaxalone depressed the electrical response to glutamate, halothane did not. In contrast, when acetylcholine was applied to the same olfactory cortical preparation, halothane and methoxyflurane stimulated the electrical response whereas pentobarbital had no effect; only alphaxalone depressed the electrical response to acetylcholine.78 Anesthetic modulation of neuronal responses to inhibitory neurotransmitters is more consistent. A wide variety of anesthetics, including barbiturates, etomidate, neurosteroids, propofol, and fluorinated volatile anesthetics, have been shown to potentiate the electrical response to exogenously applied GABA [for reviews see (60,79)]. For example, Figure 5-2 illustrates the ability of enflurane to increase both the amplitude and the duration of the current elicited by application of GABA to hippocampal neurons.80
Summary
Anesthetics alter the two fundamental determinants of neuronal communication, neuronal excitability and synaptic transmission.
Anesthetics have powerful and widespread effects on synaptic transmission that would logically contribute to general anesthesia. Thus, the synapse is generally thought to be the more relevant site of anesthetic action. Existing evidence indicates that even at the synapse, anesthetics have diverse actions, including presynaptic inhibition of neurotransmitter release, inhibition of excitatory neurotransmitter effect, and enhancement of inhibitory neurotransmitter effect. Furthermore, the synaptic effects of anesthetics differ among various anesthetic agents, neurotransmitters, and neuronal preparations.
Anesthetics have powerful and widespread effects on synaptic transmission that would logically contribute to general anesthesia. Thus, the synapse is generally thought to be the more relevant site of anesthetic action. Existing evidence indicates that even at the synapse, anesthetics have diverse actions, including presynaptic inhibition of neurotransmitter release, inhibition of excitatory neurotransmitter effect, and enhancement of inhibitory neurotransmitter effect. Furthermore, the synaptic effects of anesthetics differ among various anesthetic agents, neurotransmitters, and neuronal preparations.
Anesthetic Actions on Ion Channels
Ion channels are a likely target of anesthetic action. The advent of patch clamp techniques in the early 1980s made it possible to measure directly the currents from single ion channel proteins. Accordingly, during the 1980s and 1990s a major effort was directed at describing the effects of anesthetics on the various kinds of ion channels. The following section summarizes and distills this effort. For the purposes of this discussion, ion channels are cataloged according to the stimuli to which they respond by opening or closing (i.e., their mechanism of gating).
Anesthetic Effects on Voltage-Dependent Ion Channels
VDCCs serve to couple electrical activity to specific cellular functions. In the nervous system, VDCCs located at presynaptic terminals respond to action potentials by opening. This allows calcium to enter the cell, activating calcium-dependent secretion of neurotransmitter into the synaptic cleft. At least six types of calcium channels (designated L, N, P, Q, R, and T) have been identified on the basis of electrophysiologic properties and a larger number based on amino acid sequence similarities. N-, P-, Q-, and R-type channels, as well as some of the untitled channels, are preferentially expressed in the nervous system and are thought to play a major role in synaptic transmission. L-type calcium channels, although expressed in brain, have been best studied in their role in excitation–contraction coupling in cardiac, skeletal, and smooth muscle and are thought to be less important in synaptic transmission. The anesthetic action on L- and T-type currents has been well characterized,82,89,90 and some studies have reported the effects of anesthetics on N- and P-type currents.91,92,93 As a general rule, these studies have shown that volatile anesthetics inhibit VDCCs (50% reduction in current) at concentrations 2 to 5 times those required to produce anesthesia in humans, with less than a 20% inhibition of calcium current at clinical concentrations of anesthetics. However, some studies have found VDCCs that are extremely sensitive to anesthetics. Takenoshita and Steinbach94 reported a T-type calcium current in dorsal root ganglion neurons that was inhibited by subanesthetic concentrations of halothane. In addition, ffrench-Mullen and colleagues95 have reported a VDCC of unspecified type in guinea pig hippocampus that is inhibited by pentobarbital at concentrations identical to those required to produce anesthesia. Thus, VDCCs could well mediate some actions of general anesthetics, but their general insensitivity makes them unlikely to be major targets.
Potassium channels are the most diverse of the ion channel types and include voltage-gated, background or leak channels that open over a wide range of voltages including at the resting membrane potential of neurons, second messenger and ligand-activated, and so-called inward-rectifying channels; some channels fall into more than one category. High concentrations of both volatile anesthetics and intravenous anesthetics are required to affect significantly the function of voltage-gated K+ channels.81,96,97 Similarly, classic inward-rectifying K+ channels are relatively insensitive to sevoflurane and barbiturates.98,99,100 However, some background K+ channels are quite sensitive to volatile anesthetics.
transmembrane segments (2P/4TM; Fig. 5-3C, D).104 Patel et al.105 have studied the effects of volatile anesthetics on several members of the mammalian 2P/4TM family. They have shown that TREK-1 channels are activated by clinical concentrations of chloroform, diethyl ether, halothane, and isoflurane (Fig. 5-3B). In contrast, closely related TRAAK channels are insensitive to all the volatile anesthetics, and TASK channels are activated by halothane and isoflurane, inhibited by diethyl ether, and unaffected by chloroform. These authors further showed that the C-terminal regions of TASK and TREK-1 contain amino acids essential for anesthetic action. More recently, TREK-1 but not TASK was found to be activated by clinical concentrations of the gaseous anesthetics: Xenon, nitrous oxide, and cyclopropane.106 Thus, activation of background K+ channels in mammalian vertebrates could be an important and general mechanism through which inhalational and gaseous anesthetics regulate neuronal resting membrane potential and thereby excitability. Indeed, genetic evidence argues for a role of these channels in producing anesthesia (see later discussion).
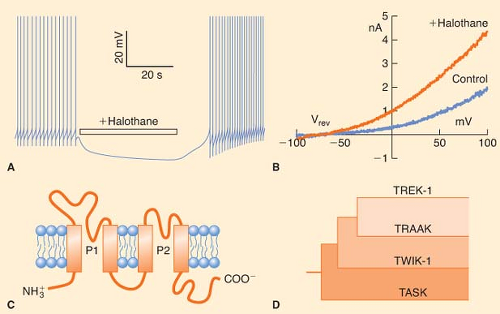 Figure 5.3. Volatile anesthetics activate background K+ channels. A: Halothane reversibly hyperpolarizes a pacemaker neuron from L. stagnalis (the pond snail) by activating IK(AN). B: Halothane (300 μM) activates human recombinant TREK-1 channels expressed in COS cells. The figure shows current–voltage relationships with reversal potential (Vrev) of −88 mV, indicative of a K+ channel. C: Predicted structure of a typical subunit of the mammalian background K+ channels. Note the four transmembrane-spanning segments (orange rectangles) and the two pore-forming domains (P1 and P2). Some but not all of these 2P/4TM K+ channels are activated by volatile anesthetics. D: Phylogenetic tree for the 2P/4TM family. (Reproduced with permission from Franks NP, Lieb WR: Background K+ channels: An important target for anesthetics? Nat Neurosci. 1999;2:395.) |
Volatile anesthetics, propofol, and ketamine have been shown to inhibit HCN-mediated currents in both cell culture and native mouse neurons. The Bayliss group has shown that both halothane and isoflurane shifted activation of HCN1 channels expressed in cultured cells to a more negative membrane potential and reduced current amplitude.108 In isolated spinal motor neurons, halothane reduced the Ih current, consistent with the inhibition of HCN channels. Similar inhibition of HCN1 channels was subsequently observed with clinical concentrations of propofol and ketamine.109,110 The inhibition of HCN1 by ketamine was stereoselective in the same manner as its stereoselectivity for general anesthesia.109 Notably, in this same study, etomidate was not found to inhibit HCN1 channel activation. Thus, HCN1 channels may be important for the actions of both volatile anesthetics and a subset of intravenous anesthetics. Genetic experiments described below argue that anesthetic inhibition of the HCN1 channel may contribute to ketamine and propofol anesthesia in vivo.
Summary
Existing evidence suggests that most voltage-dependent ion channels are modestly sensitive or insensitive to anesthetics. However, some sodium channel subtypes are inhibited by volatile anesthetics and this effect may be responsible in part for a reduction in neurotransmitter release at some synapses. Additional experimental data will be required to establish whether anesthetic-sensitive voltage-dependent ion channels are localized to specific synapses at which anesthetics have been shown to inhibit neurotransmitter
release. Recent evidence suggests that members of the 2P/4TM family of background potassium channels may be important in producing some components of the anesthetic state. In addition, the HCN family of channels has emerged as a potentially relevant anesthetic target for both volatile and intravenous anesthetics.
release. Recent evidence suggests that members of the 2P/4TM family of background potassium channels may be important in producing some components of the anesthetic state. In addition, the HCN family of channels has emerged as a potentially relevant anesthetic target for both volatile and intravenous anesthetics.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


