Mechanical Ventilation in the Acute Respiratory Distress Syndrome: Introduction
Few areas of critical care medicine have been the subject of as much investigative attention or clinical concern as the set of problems grouped under the label acute respiratory distress syndrome (ARDS). This syndrome, first formally described in 1967,1 continues to be recognized clinically as a rapidly developing impairment of pulmonary oxygen exchange accompanied by diffuse infiltrates and altered respiratory system mechanics that cannot be attributed solely to hydrostatic forces. Fueled by better characterization of innate pathophysiology and of iatrogenic factors, considerable progress has been made in recent years toward reducing the adverse consequences of this condition. Yet, after more than four decades, active debate continues regarding key elements of the ventilatory prescription and appropriate therapeutic targets.
From the outset, mechanical ventilation with positive pressure has been essential in addressing the life-threatening gas exchange abnormalities and otherwise unsustainable workloads associated with ALI. Only in the relatively recent past, however, has there been clear documentation that the tidal pressures of mechanical ventilation can impact morbidity and survival.2,3 This awareness has caused a conceptual shift away from attempting to restore normal blood gases at the costs of high pressure and toward adopting the avoidance of preventable iatrogenicity (“lung protection”) as the first priority.
Many aspects of the debate concerning appropriate ventilator management of this group of conditions can be traced to the heterogeneity of the patient population, to our still imperfect comprehension of the mechanisms of ventilator-associated lung injury (VILI) and to the relative imprecision of the criteria upon which the label ARDS and/or acute lung injury (ALI) is assigned.4 Despite an incomplete and still evolving understanding, a rich experimental and clinical database—much of it collected over the past two decades—allows for the development of a rational set of principles upon which to formulate an effective ventilation strategy.5–7 Definitive answers for many important clinical questions related to this topic are not available; what is presented here reflects a pathophysiology-guided approach to accomplish essential clinical objectives while avoiding VILI (Table 29-1).
|
General Objectives for Ventilator Support
In the clinical setting, mechanical ventilation ensures adequate oxygenation of arterial blood, provides sufficient oxygen transport to vital organs and tissues, assists in eliminating carbon dioxide, relieves excessive burdens placed upon the respiratory muscles, helps maintain alveolar stability, and allows therapeutic measures that require controlled ventilation. Despite its undeniable value, however, mechanical ventilation also has the potential to inflict adverse clinical outcomes. The task of accomplishing ventilation safely in patients with injured lungs is made far more difficult by the mechanical heterogeneity of the respiratory system and the diversity of pathophysiology encountered among different patients who satisfy extant operational criteria for this condition.
Defining the Problem
The terms ALI and ARDS comprise a category of patients with varied pathoanatomy and mechanical characteristics. According to the widely used American-European consensus guideline,4 the primary criteria relate to pulmonary oxygen exchange, the appearance of the plain chest radiograph, and to a clinical assessment of left ventricular function. Although such broadly inclusive criteria may be useful for some purposes, they prove problematic for others. In formal definitions, for example, no provision is made for the level of positive end-expiratory pressure (PEEP) at which the arterial sample is obtained or the chest radiograph is exposed. No stipulation requires that the defining criteria be met under standardized conditions and remain reproducible over time. Chest wall properties and body weight are left unaccounted for. Yet, pulmonary oxygen exchange is influenced not only by the properties of the lung, but also by end-expiratory lung volume, body position, mixed venous oxygen content, pulmonary blood flow, and the integrity and intensity of hypoxic pulmonary constriction. The chest radiograph is interpreted subjectively, and expert assessments often differ.8 Moreover, it is clear that the lungs of different patients with ARDS vary with regard to radiographic appearance, inherent recruitability, and histopathology. Even within the same individual, assessed at the same moment, the pathoanatomy and mechanical environment varies from site to site within the injured lung. Such regional differences are partially explained by the properties of the surrounding chest wall, which profoundly influence the inflation characteristics of the integrated respiratory system as well as their regional distribution.9,10
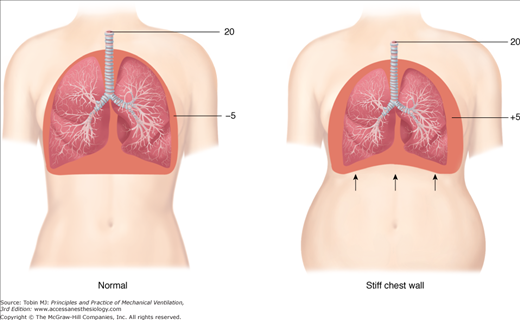
Similar clinical presentations can mask radical differences in lung pathology, mechanical properties, and response to ventilator settings and maneuvers. Lungs of patients with ARDS resulting from pneumonic consolidation, for example, are less likely to inflate easily than are lungs made edematous by the circulating mediators of extrapulmonary sepsis.11 Computed tomographic (CT) patterns may reflect these differences.12 Moreover, inflexibility of the chest wall may substantially increase the pressures required to inflate the respiratory system (Fig. 29-1).13 From these considerations, it is clear that inflexible guidelines for selecting PEEP and tidal volume that advise specific numerical values of these settings will be variably effective in accomplishing the goal of minimizing tissue stresses, depending on the type and severity of lung injury, the compliance of the chest wall, and the ranges for opening pressures and closing tendencies among the multiplicity of the lung units that comprise the injured lung.
Pathophysiologic Features Relevant to Ventilatory Support
A major conceptual advance of the past 25 years is the recognition that the lungs of patients with ARDS are mechanically heterogeneous and vary enormously in their patterns of infiltration, their inflation and collapse properties, and their regional expressions of pathology. CT scanning has revealed densities that may be localized or diffuse—with implications for response to ventilator interventions such as PEEP.12 Variation in the underlying conditions that give rise to the clinical problem described as ARDS precludes categorical histologic descriptions that apply across the full range of clinical experience. A few salient characteristics, however, are shared by most. In its earliest phase, noncardiogenic (high permeability) edema gives rise to a lung whose parenchymal airspace is partially occupied by proteinaceous edema and cellular infiltrate. A relatively large proportion of the lung—often exceeding 50%—is airless at end expiration, with the exact percentage depending jointly on severity, disease stage, etiology, and PEEP. Destruction of surfactant-producing type 2 alveolar cells leads to its diminished production, while exuded proteins and inflammatory products compromise the viability of the surfactant that remains.14 Surfactant plays several important biologic and physiologic roles. From a purely mechanical standpoint, the loss of functional surfactant increases surface tension, thereby contributing to alveolar flooding, increased tissue elastance, small airway closure and atelectasis—particularly at low lung volumes.
The relative proportions of airless and aerated tissue also vary with disease type and stage. Although some controversy persists, CT scan estimates of gas and tissue volumes suggest that potentially “recruitable” tissue comprises only a minority of radiographic density at functional residual capacity in most cases.15 Flooded and consolidated lung units comprise the remainder. Although clearly a minority viewpoint, a plausible argument has been advanced that “reopening” of lung units by alveolar pressure may occur primarily by redistributing alveolar liquid and shifting fluid volume from the alveolar to the interstitial compartments of the lung—not by atelectasis reversal (“recruitment”).16 The rapidity with which CT tissue density develops and resolves, however, as changes of alveolar pressure are imposed, as well as direct observations by intravital microscopy of alveoli at the lung’s surface,17 cast doubt on the primacy of the “fluid-shift hypothesis.”
Replacement of airspaces by inflammatory debris, cells, and fluid results in a lung whose aeratable capacity and compliance (measured in terms of volume accepted per unit of pressure) is severely reduced. The compliance of the respiratory system (lungs and chest wall) falls during ARDS for two reasons. First, airspace is lost to fluid and cellular infiltrates. Second, many functional lung units operate near their elastic limit because many fewer are available to accept the tidal volume. Under normal conditions, surfactant-modified surface forces allow the lung to inflate and deflate at similar pressures. In contrast, the injured lung is characterized by a right-shifted pressure volume loop, made so by its reduced aeratable capacity and by surfactant depletion.13,18,19 For the same tidal volume, therefore, the mechanical work of breathing is dramatically increased.
Although clinicians characterize the mechanics of the injured lung by airway pressure and flow measurements made at the airway opening—the common entry and exit point for gas exchange with the environment—the mechanical properties of the lung’s individual subunits are hardly uniform, even in health. In part, this heterogeneity relates to regional variations of pleural (and therefore transpulmonary) pressure that arise from interactions of the chest wall with the injured lung and from the need for dependent tissues to support the weight of the mediastinal contents and the edematous lung (Fig. 29-2). This underlying mechanical heterogeneity is implied by quantitative CT imaging techniques that characterize the topographical anatomy in response to changing patterns of airway pressure, or more directly in real time by imaging of ventilation with radiotracer gases or electrical impedance tomography.20,21
Figure 29-2
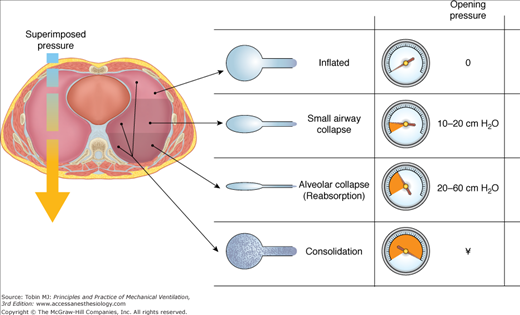
Spectrum of opening pressures associated with lung units within the injured lung. Lung units located in dependent areas are compressed by the weight of the overlying lung and mediastinum. Local transpulmonary pressures vary considerably, so that some lung units remain fully inflated throughout the tidal cycle, whereas others cannot be aerated. Less pressure is required to open small airways when the alveoli remain aerated than when all alveolar gas has been absorbed. (Image provided by Luciano Gattinoni and used with permission.)
For any specified airway pressure, some lung units are closed and others are open. Among the population of open units, there exists a range of states of lung unit expansion, depending on the local transpulmonary pressures that distend them. Even at airway pressures that are generally considered modest, some of the open lung units verge on overdistension, while others are on the compliant portion of their pressure volume relationship (Fig. 29-3). Alveolar distension that approaches the elastic limit stimulates surfactant production, but the repetitive application of nonphysiologic stretching forces, as during high tidal volume or high-pressure tidal ventilation, initiates molecular signaling of a local inflammatory response.22,23
Figure 29-3
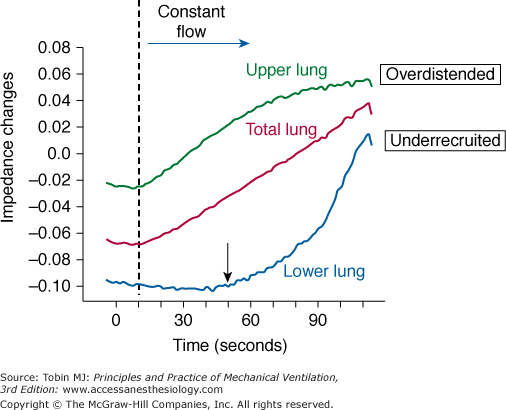
Mechanical characteristics of lung units in dependent (lower) and nondependent (upper) lung regions. As the lung is inflated by constant flow of gas, electrical impedance increases in proportion to aerated volume. Although the relationship of volume to impedance for the total lung appears roughly linear, upper lung regions are relatively overdistended whereas lower lung regions are relatively underrecruited until total lung capacity is achieved. (Image provided by Marcelo Amato and used with permission.)
Mechanical interdependence among the lung units of a heterogeneously affected lung amplifies the stresses of high pressure at the junctions of closed and open lung units in a nonlinear fashion (Fig. 29-4).23,24 Such recurring forces at the points of stress focusing not only strain the lung’s structural meshwork to initiate inflammation, but also assist in reopening potentially recruitable (atelectatic) lung units.25 When atelectatic tissues are subjected to high pressures, amplified shearing forces within these zones at high risk for damage may be of sufficient magnitude to tear the delicate terminal airways or alveoli themselves, creating micro wounds that produce tissue hemorrhage and incite inflammation as a secondary phenomenon. Stress amplification at the margins of dissimilar tissues is at least partially a function of their relative volumes. (A flooded, gasless alveolus of volume similar to its air-filled neighbors may experience forces that are no more intense.) Because mechanical forces are amplified at the junctions of dissimilar tissues, it seems likely that injury abets the process of ventilator associated injury, so that damage propagates outward from injured into previously healthy zones. Whether injury results from the process of repetitive popping open of lung units under high pressure or simply is the cumulative result of high tension stretching at the interface remains to be resolved. It appears, therefore, that there are at least two critical elements that place the injured lung at risk for ventilation associated lung damage: high inflating pressures and the prevalence of collapsed lung units that interface with open ones.
Figure 29-4
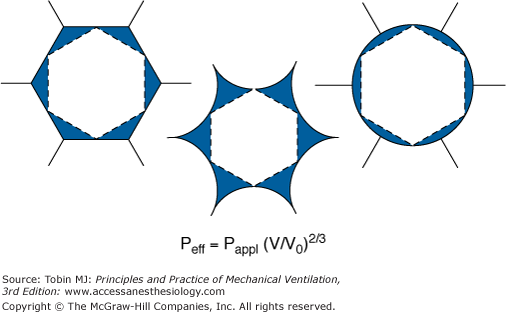
Amplification of tension at the boundaries of open end collapsing lung tissue. Normally distended (left), collapsing (center), and overdistended (right) states of inflation. Because of interdependence, the tensions applied to a collapsing alveolus are amplified. A simple mathematical model predicts that the tensions resulting from an alveolar pressure (Pappl) produce a tension comparable to an effective pressure (Peff) described by the equation. At 30 cm H2O applied pressure, the volume of an aerated alveolus (V) is approximately ten times that of a collapsed alveolus (V0). Thus, the estimated pressure necessary to mimic the tensions at the interface is approximately 4.5 times as great as that applied, or 140 cm H2O. (Used, with permission, from Mead et al.23)
At the microscopic level, lung unit collapse may be thought of in two broad categories: “loose” atelectasis that arises primarily from compressive forces of the heavy lung acting to close small airways and responds to relatively low levels of transpulmonary pressure, and adhesive or “sticky” atelectasis that results from gas absorption and requires very elevated pressures to reverse.26 High concentrations of inspired oxygen encourage the latter in poorly ventilated regions, as well as interfere with surfactant kinetics. These hard to recruit units may be among the first to close as high airway pressure is withdrawn. Loose and sticky types of atelectasis often coexist, perhaps accounting for the fact that lung units open throughout the entire range of total lung capacity (Fig. 29-5).25–27 In the setting of severe ARDS, pressures that exceed 60 cm H2O may be needed to complete the recruiting process. Although opening of lung units is primarily a function of transalveolar pressure, the duration with which pressure is applied also contributes; that is, a lower pressure may be sufficient if applied for an adequate period. Moreover, atelectatic lung units tend to “yield” (open) in a stuttering, discontinuous fashion, with high sustained pressure causing a serial snapping open of small blocks of units.28 Discontinuous lung opening during the tidal cycle may give rise to audible crackles, usually best heard in dependent regions.
Figure 29-5
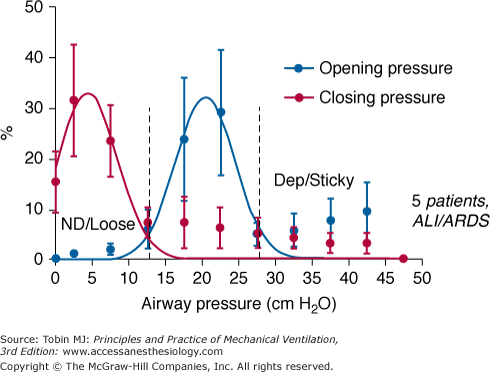
Histograms relating percentage of potentially recruitable lung units that are open to inflation and deflation airway pressures. Unstable airways open at relatively modest pressures, whereas alveoli that have undergone absorption collapse require much higher pressures to open. The spectrum of closing pressures is left shifted. Note that some lung units begin to collapse at relatively high airway pressures. (Reprinted with permission of the American Thoracic Society. Copyright © 2012 American Thoracic Society. Crotti S, Mascheroni D, Caironi P, Pelosi P, Ronzoni G, Mondino M, Marini JJ, Gattinoni L. Recruitment and derecruitment during acute respiratory failure. A clinical study. Am J Resp Crit Care Med. 2001;164(1):131–140. Official Journal of the American Thoracic Society. Adapted from original article.)
The increased microvascular permeability of ALI and/or ARDS renders it vulnerable to edema formation and impedes its relative rate of edema clearance. The weight of the normal lung does not begin to rise significantly until pulmonary venous pressure exceeds 20 mm Hg. Unlike the healthy lung, however, extravascular water in the injured lung bears a strong quasilinear dependence on hydrostatic microvascular pressure, a relationship that begins from capillary pressures that are considered low by most clinical standards.29 The term noncardiogenic pulmonary edema, therefore, does not imply that gas exchange cannot improve by lowering the hydrostatic pressure gradient across the injured lung. Because extraalveolar microvessels as well as capillaries can leak,30,31 numerous factors influence the vascular pressures relevant to edema formation. These include left ventricular filling pressure, cardiac output (which increases the pressure needed to drive blood flow through the lung with limited capillary recruiting reserve), the plasma oncotic pressure, the interstitial fluid pressure, and the integrity of the alveolar and lymphatic lung water clearance mechanisms. Clearance mechanisms are influenced by the degree of lung stretch. Translocation of blood from infradiaphragmatic vascular capacitance beds to the central vessels of the thorax can occur as intraabdominal pressure rises.32
Mechanisms and Consequences of Ventilator-Induced Lung Injury
For more than three decades, experimental studies have shown that excessive mechanical stresses developed during mechanical ventilation can inflict injury upon both normal and acutely injured lungs, and once injured, retard their healing.33,34 Injury may result from overstretching of tissues that are already open, from shearing forces at the junctions of expandable and unyielding tissue, or from repeated percussion of closed terminal airways (Fig. 29-6). Repeated application of transalveolar pressures exceeding those corresponding to the inflation capacity of a healthy lung may disrupt the alveolar epithelial barrier, especially in the absence of sufficient end-expiratory pressure to reduce stress focusing by holding open mechanically unstable lung units.33,35 From a theoretical standpoint, sustained recruitment reduces the potential for damaging forces to concentrate at the boundaries of inflating lung and unyielding or collapsed structures. Indeed, an overwhelming body of experimental work indicates the lung protective effect of sustained “recruitment” when high tidal inflation pressures are used.36,37 Mechanosignaling at moderately high airway pressures may induce the formation and release of inflammatory mediators,22,37–42 initiate programmed cell death (apoptosis), or produce necrosis.38,41 Excessive strain may exceed cytoskeletal tolerances, causing physical tears and stress fractures of endothelium, epithelium, and intercellular matrix (Fig. 29-7).38,39 Scientific data suggest that when the ratio between tidal volume and aerated functional residual capacity exceeds 2, the resulting transalveolar forces are sufficient to cause tissue damage.40 Furthermore, the tendency for translocation of inflammatory mediators and bacteria into the bloodstream is influenced by the ventilator pattern.42–44 Such observations suggest possible links between adverse patterns of ventilation and dysfunction in remote vital organs.
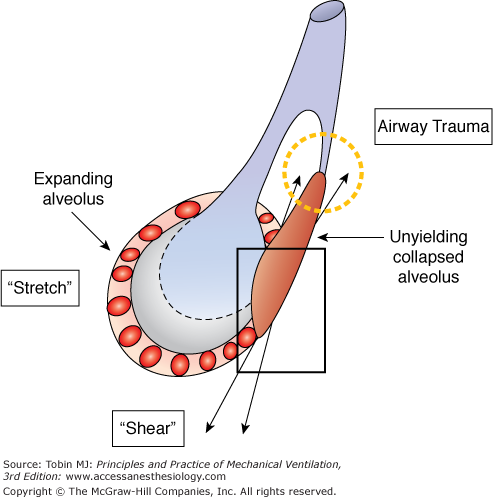

From available data, VILI appears to be a complex process initiated by the repetitive application of excessive, stress/strain to the lung’s fibroskeleton, microvasculature, terminal airways, and delicate juxtaalveolar tissues. Defining the linkage between stress, strain, and diffuse alveolar damage is currently a subject of intense investigation.22,37,38,42,43,45 It seems undeniable, however, that high levels of mechanical stress disrupt the normal functioning of cells that populate the pulmonary microenvironment and that sufficient dimensional strain triggers the release of inflammatory mediators and destructive enzymes (Fig. 29-8).43,46,47 Under moderate degrees of strain, such mechanosignaling may be the primary pathway to injury. When the applied mechanical stress is very high, fibroelastic structural integrity may be directly breached, with the inflammatory process a consequence rather than initiator of the observed histopathology. These high tissue-rupturing forces are especially likely to be generated in dependent lung regions, where unstable alveoli are most prevalent (Fig. 29-9). Another important mechanism in the causation of lung tissue damage may occur at the level of the terminal airways as they remain closed as airway pressures build or open and reclose with each tidal cycle.36 Damaging shear forces (those that run tangential to the structure) appear to rip the epithelium from its attachments. The rate at which these tissues expand, conditioned by the inspiratory flow profile, may also play an important role.48 Although relatively few studies have focused on this aspect, such a contribution seems likely, as the expansion velocity of the parenchyma is amplified by the reduced number of functioning airways that must accept what is typically an elevated total minute ventilation.
Figure 29-8
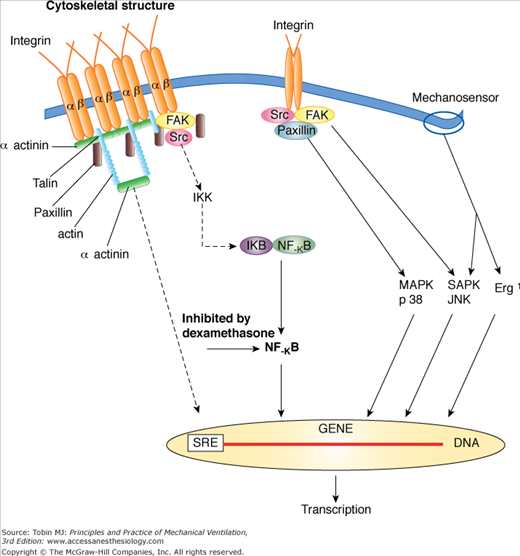
Mechanosignaling pathways of inflammation under conditions of excessive tissue strain. (Used, with permission, from Dos Santos and Slutsky.37)
Figure 29-9
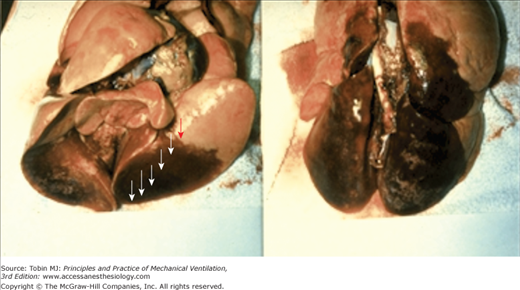
Excised heart–lung block from a previously normal animal subjected to high inflation pressure and low levels of positive end-expiratory pressure (PEEP). Specimen is shown inflated at 20 cm H2O airway pressure in the supine (left panel) and prone (right panel) conditions. Note the sharp demarcation of hemorrhagic edema from relatively normal appearing lung. Damage may progress sequentially from dependent to nondependent regions, as indicated by the sequence of arrows.
From an engineering perspective, mechanical stress is a function of transstructural tension; strain is the dimension-altering consequence of high transstructural pressure, conditioned by the elastance of the element in question. Although by no means the only determinant of tissue forces, the measurable analog of the stress across the entire lung is transpulmonary pressure—crudely estimated as the difference between static airway pressure (the “plateau” pressure) and average pleural pressure (often estimated by use of an esophageal balloon).49 Regional transpulmonary pressures vary considerably, because of the influence of gravity, chest wall irregularities, intraabdominal pressure, mediastinal weight, and vascular-filling pressure (see Fig. 29-2).50,51 Available laboratory data illustrate that pleural pressure, however measured, does not accurately reflect interstitial pressure when the lung is edematous and inflamed.51,52 Even directional changes of pleural and interstitial pressures may not track together in a nonuniform environment (Fig. 29-10). Although not directly measurable, strain correlates with aerated volume as a fraction of aeratable capacity. As already noted, excessive strain may be experienced when VT exceeds the aerated functional residual capacity of a well-recruited lung by a factor of 2.040 (Fig. 29-11).
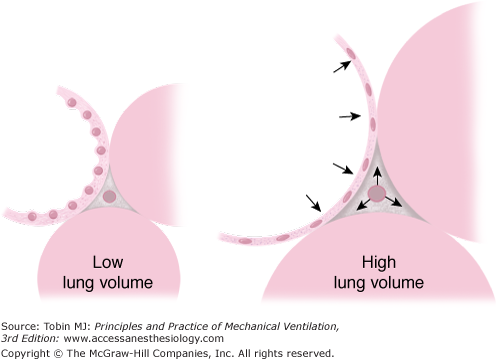
Figure 29-11
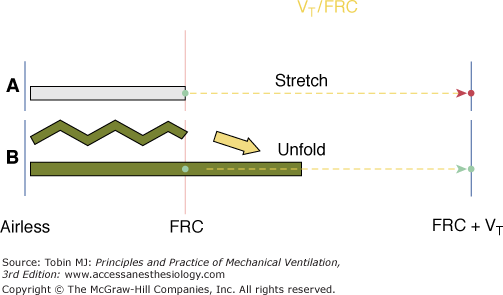
Concept of strain as relates to tidal volume (VT) and functional residual capacity (FRC). Assuming that the lung has few recruitable units at rest (A), doubling of FRC by the applied VT will result in excessive tissue strain (VT/FRC ≥2). Peak transalveolar pressure would reflect that damaging stress. If tidal recruitment occurs during delivery of the VT (B), the recruited tissue will relieve the stress even though the calculated ratio of VT to FRC initially is the same. Peak transalveolar pressure would not rise as high as in situation A, but depending on driving pressure amplitude, tidal recruitment might have potential to injure secondary to stress amplification.
Once this latter concept is understood, it becomes clear that tissue stress is not a predictable function of tidal volume but rather is strongly influenced by tidal volume in relation to the size (and compliance) of the compartment forced to accept it. Logically, the developed transpulmonary pressure would seem a variable that integrates these relationships. Even this, however, is not the full story—stress amplification and flow-influenced shearing forces at the boundaries of tissues with differing capacities to expand must be considered. Peak tidal transalveolar pressure interacts with end-expiratory transalveolar pressure to determine measurable stress, which varies markedly from site to site across the damaged lung. The alveolar pressures resulting from tidal volume and PEEP are both interactive elements in the injury process, but even when considered together cannot precisely estimate tissue stresses, as interstitial pressure is an unmeasured variable that differs throughout.
Moreover, the damaging strain that results from a given stress depends on tissue fragility. In the setting of ARDS, tissue integrity is likely to be at least normally delicate or even compromised, especially in its earliest phase. The low compliance typically associated with ALI results primarily from the reduced number of aeratable lung units—not from increased stiffness of the individual lung units themselves.26,52 The tidal volumes pulled during exercise by a healthy athlete may exceed 25 to 30 mL/kg of lean body weight, whereas much smaller breaths eventually cause damage or lung rupture in the setting of ALI. Although increasing the tidal volume delivered to the same individual will invariably cause tissue stresses to rise in some lung units, a raised tidal volume may actually cause the stresses in other units to fall. This apparent paradox owes to the recruiting effect of the higher pressures and perhaps improved ventilation of marginally ventilated units predisposed to absorption collapse.53 At any clinically selected tidal volume, tissue stresses may be reduced or accentuated by the application of PEEP, depending on whether PEEP effectively recruits vulnerable tissue or simply raises plateau pressure. Quests for a universally applicable predetermined tidal volume or PEEP value applicable to all patients would seem destined to be a fruitless exercise.
Modifying the characteristics of the chest wall (e.g., by prone positioning54,55) is an effective mechanism for altering the regional distribution and amplitude of transpulmonary pressure. Even within the same small region, inflationary stresses can vary markedly in magnitude and even in direction between structures situated within microns of one another. Shearing forces, one of the varied forms of mechanical stress resulting from lung inflation, intensify at the junctions of tissues with different compliance values and anchoring attachments.23 Minimizing or eliminating these irregularities reduces the potential for adverse “stress focusing” and tissue strain. In such a microenvironment, limiting end-tidal alveolar pressure assumes major importance for two primary reasons: (a) A high plateau pressure may overstretch open alveoli, and (b) perhaps more importantly, because junctional tension rises in nonlinear proportion to airway pressure,23 the plateau pressure acts as a potent lever arm at stress focus points.
By reducing the number of junctional interfaces and by preventing the repetitive opening of terminal lung units at high pressure, recruitment of lung tissue—defined as the sustained reversal of atelectasis on whatever scale it occurs—may be lung protective. When nearly all potentially recruitable tissue is aerated, the lung is said to be “open.”56 A given transpulmonary pressure applied to a fully open lung should be associated with less stress than the same pressure applied to a lung with closed units juxtaposed to open ones (Fig. 29-12). Some authors argue that the injured lung should be fully “opened” so as to reduce the potential for repetitive opening and reclosure.56 Presently, however, it is not clear that this should always be given highest priority; to what extent repetitive opening and closure of small airways produces injury and whether the prevention of such behavior is the key to lung protection remain debatable, especially when peak tidal pressures are restrained; modest airway pressures may not inflict shearing injury when opening occurs at low pressure. In addition, because the difference between opening and closing pressures is often quite narrow in units that do open at higher pressure, some degree of tidal recruitment may be unavoidable. Therefore, how much of the lung should be “opened” and what pressure cost is acceptable remain key unresolved questions.
Figure 29-12
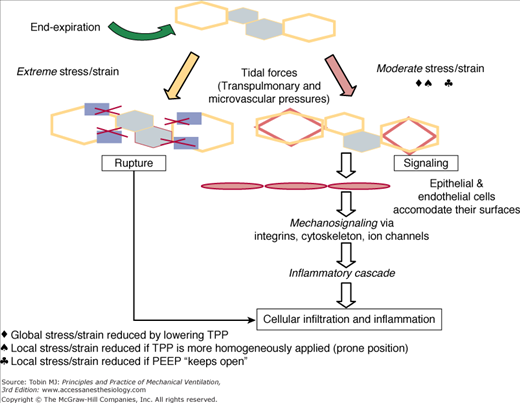
Hypothetical relationship between tissue damage and the severity of mechanical stress or strain during the tidal cycle. Moderate forces applied repeatedly to junctional tissues may result in mechanosignaling of inflammation. Extreme stress or strain causes micro wounds to develop, with inflammation occurring as an epiphenomenon. PEEP, positive end-expiratory pressure; TPP, trans-pulmonary pressure. (Used, with permission, from Marini JJ, Gattinoni L. Ventilatory management of ARDS: a consensus of two. Crit Care Med. 2004;32(1):250–255.)
Experimentally, a variety of cofactors apart from end-inspiratory, end-expiratory, and driving tidal transpulmonary pressures are important in the generation or prevention of VILI (Table 29-2). Prone positioning confers a protective advantage in both normal and preinjured animals.57,58 A potential role for high inspired oxygen fraction as a contributor to iatrogenic injury has been intimated for many years but never convincingly documented in the clinical setting.1,37 As other examples, higher precapillary59 and lower postcapillary60 vascular pressures intensify the injury caused by a fixed ventilatory pattern. As the lung expands, alveolar and extra-alveolar microvessels are compressed and dilated, respectively (see Fig. 29-10). At some intermediate point on the luminal pathway that links them, the pushes and pulls are oppositely directed, giving rise to forces that stress the microvascular endothelium. Energy dissipation across the waterfall created by intermittent zone 2 conditions, vascular interdependence, and opening and closure of the microvascular endothelium could potentially explain the damaging influence of reduced postalveolar vascular pressure.
|
For identical tidal inflation and end-expiratory pressures, reducing respiratory frequency and minute ventilation attenuates or delays damage, provided that the tidal ventilatory stresses are sufficiently high.61 Large tidal breaths and sighs occur infrequently as part of an inherent pattern of normal breathing. Sighs, for example, are inherent to the natural breathing pattern and are not injurious.62 Multiple high-tension tidal cycles are required to signal inflammation. Experimental evidence also demonstrates that effective repair of minor defects within the cell membrane may occur within seconds of reducing stress,63,64 and reducing respiratory frequency may allow these repair processes sufficient time to run to completion. Alternatively, less cumulative damage to the lung may occur per unit time as injurious forces unzip structural elements of the lung’s fibroskeleton.
What level of transpulmonary pressure is likely to be damaging, therefore, depends on variables other than the tidal “plateau” pressure. Moreover, when the lung is comprised of large numbers of recruitable units, PEEP attenuates the tendency for high plateau pressures or tidal volumes to cause injury.65–67 Consequently, it is difficult to specify an exact level of transpulmonary pressure that serves as an appropriate threshold criterion for safety. From a theoretical standpoint, repeated tidal application of a transpulmonary pressure of 15 to 20 cm H2O (corresponding in a patient with a normal chest wall to a plateau pressure that may be in the range of 25 to 35 cm H2O) gives cause for concern, as some higher compliance regions of the injured lung may approach their elastic limits at this pressure. It is worth noting that a transpulmonary pressure of only 15 cm H2O subjects the normal lung to about two-thirds of its total capacity and is associated with a tidal volume exceeding 2500 mL.13
Experimental data suggest that lung tissues can be injured by mechanical stretching forces more easily in the setting of other noxious influences. In other words, it takes a higher stretching force to cause injury in a previously healthy lung than in one that has been exposed previously or concurrently to another inflammatory stimulus, such as endotoxin, hyperoxia, or cytotoxic drugs.68,69 Such observations have given rise to the “two-hit hypothesis” for VILI, and underscore the potential vulnerability of the preinjured lung to imprudent ventilatory prescriptions. At the same time, such vulnerability can be viewed as an opportunity to modulate the severity of VILI by altering both nonventilatory and ventilatory factors.
A post hoc analysis of the ARDS Network tidal volume trial (ARMA) indicates that observed mortality paralleled plateau pressure to very low values—considerably lower than 20 cm H2O (Fig. 29-13).70 This correlation, which is difficult to explain entirely by indices of disease severity, suggests the possibility that for the injured lung there is no obvious safety threshold below which ventilator-associated lung damage does not occur. Other data suggest danger at relatively low plateau pressures,71 but detailed analysis of human clinical trials argue that plateau pressures less than 30 cm H2O have little impact on mortality once PEEP and disease severity are accounted for.72 (Tidal volume must correlate with plateau pressure in any given individual; prestudy tidal volume and plateau pressure data from the ARMA trial, however, showed remarkably poor correlation because compliance [the inverse of elastance] is the missing parameter [Fig. 29-14].)
Figure 29-13
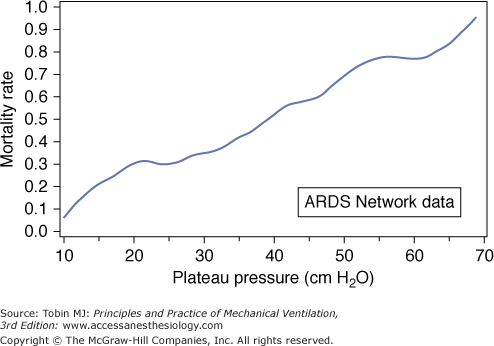
Relationship of mortality rate to plateau pressure generated during the National Institutes of Health-sponsored ARDS Network multicenter trial of high versus low tidal volumes. Note the quasilinear relationship of mortality rate to plateau pressure. (Used, with permission, from Brower et al.70)
Figure 29-14
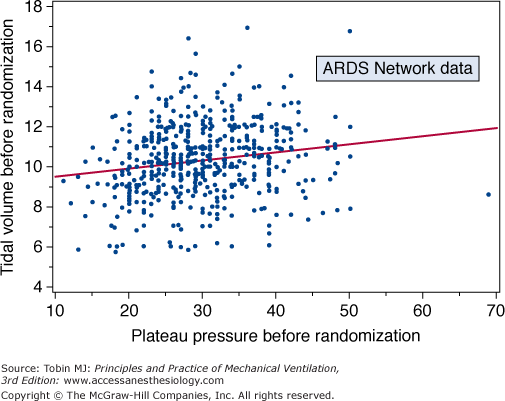
Scatterplot of tidal volume versus plateau pressure before randomization to the limbs of the National Institutes of Health-ARDS Network ARMA trial. (Used, with permission, from Brower et al.70)
Elevating the pressure baseline from which breaths are taken or delivered raises mean and end-expiratory lung volumes. Doing so nearly always improves oxygenation to some extent—primarily a function of keeping the lung open. When the breaths are drawn spontaneously or with pressure support, it is customary to call the end-expiratory pressure continuous positive airway pressure (CPAP); during breaths of predetermined length, the term is PEEP. Both are instrumental in the supportive care of patients with ARDS.
The potential clinical utility of PEEP in improving oxygenation was mentioned in the paper that first brought ARDS to widespread clinical attention.1 Subsequent work demonstrated its potential to increase ventilation dead space and impair cardiac output by impeded venous return and increased right ventricular afterload.73 In patients in whom PEEP stabilizes lung units that are susceptible to collapse, the response to increasing PEEP is generally to improve pulmonary oxygen exchange efficiency. Many patients show limited or negligible response, however, presumably because the recruitable population of lung units under baseline conditions is small. Infrequently, increasing PEEP can actually cause the partial pressure of arterial oxygen (PaO2) to fall, presumably by redirecting pulmonary blood flow, by causing pulmonary arterial pressure to rise high enough to shunt venous blood through a patent foramen ovale, or by causing sufficient reduction in oxygen delivery to force an increase in systemic O2 extraction and desaturate venous blood (see Chapter 37).
The benefit of PEEP on oxygenation depends on improving functional residual capacity. When patients labor to breathe, as during hyperpnea, expiratory efforts made against PEEP may force the lung’s volume to fall below its equilibrium value, effectively storing elastic energy to aid inspiration but interfering with improvement in O2 exchange. Under these circumstances, eliminating forceful expiratory muscle action (as by sedation) tends to improve O2 exchange efficiency.74
Because PEEP has the potential to maintain recruitment of unstable lung units (thereby reducing “stress amplification”), there has been intense interest in its role as a core element of a lung protective ventilating strategy. When plateau pressure is held constant, raising PEEP not only reduces the number of closed lung units, but also shortens the lever arm (“driving pressure”) applied to the unstable lung units at risk to open forcefully (Fig. 29-15). Yet, when tidal driving pressure is preserved, PEEP raises both mean and peak tidal pressures, distends lung units that are already open, redirects blood flow, and alters cardiac loading conditions. Moreover, those lung units at the interface of closed and open units that remain unopened or continue to undergo repeated cycles of tidal recruitment despite an increase of PEEP are subjected to any PEEP-related elevation of end-inspiratory pressure, increasing the tendency for damage to those specific units. Thus, PEEP has the clear potential for benefit or harm depending on the balance among its multiple effects. Prone positioning tends to even the distribution of ventilation and reduce the gradient of transpulmonary pressure across the lung that exists in the supine position.55,75 Airway and lymphatic drainage also tend to improve. This improved uniformity facilitates the selection of a single combination of PEEP and tidal volume that achieves a protective strategy for the entire organ.
Figure 29-15
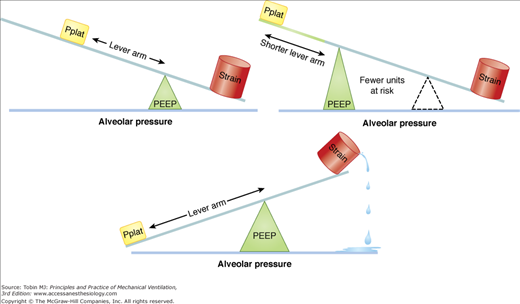
Hypothetical relationship among plateau pressure PEEP and tissue strain. In the generation tissue strain, the driving pressure for tidal volume (the difference between plateau pressure and PEEP) acts as a force lever arm, whereas PEEP is the fulcrum. Upper Left. Levels of plateau pressure (Pplat) and PEEP produce strain within acceptable limits. Bottom Center. Increasing Pplat while keeping PEEP at a level insufficient to hold open unstable units lengthens the lever arm and produces excessive tissue strain. Upper Right. When PEEP is increased, the same high Pplat that caused damage previously is better tolerated in that fewer lung units are placed at risk after recruitment and the lever arm of driving pressure is shortened.
A major clinical challenge is to apply sufficient pressure to keep the lung fully recruited without either increasing the stress applied to tissue that remains closed or overdistending alveoli that remain patent. Certain steps can be taken to minimize the number of unstable units by reversing those conditions that predispose to compressive and absorptive atelectasis (Table 29-3). PEEP cannot keep open lung units that were never open, and the recruiting process is not completed until pressures are reached that considerably exceed the total capacity of the healthy lung (Fig. 29-16). Although most lung units open at pressures less than 25 cm H2O, some refractory units of the acutely injured lung may require much higher pressures to establish patency. To reach the “yield” (opening) pressures of refractory lung units requires the initial application of pressures that would be hazardous during tidal ventilation.15,27,76 Newly opened lung units tend to close at lower pressures, allowing ventilation with the same tidal volume and PEEP to occur in the context of a more open lung with fewer lung units at risk for opening and closure (Fig. 29-17).
Figure 29-16
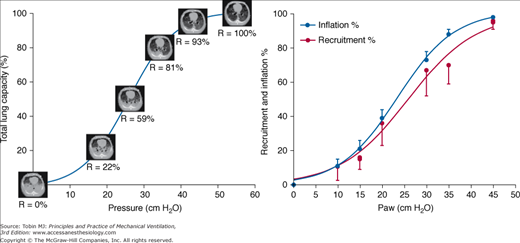
Left. CT scan slices obtained after oleic acid injury of the lung superimposed on the pressure-volume relationship of the respiratory system. Consolidation and radiographic density are greatest in the dependent lung regions and high pressures must be applied, even in this “highly recruitable” lung model, to fully reverse the radiographic evidence for collapse. Percentages denote aerated recruitable tissue. Right. Recruitment and inflation percentages as functions of static airway pressure. In these five patients with ARDS, the pressure–recruitment curve parallels the pressure–inflation curve when both are expressed as percentages of their maximum ranges. (Used, with permission, from Pelosi et al27 and Crotti et al.15)
Figure 29-17
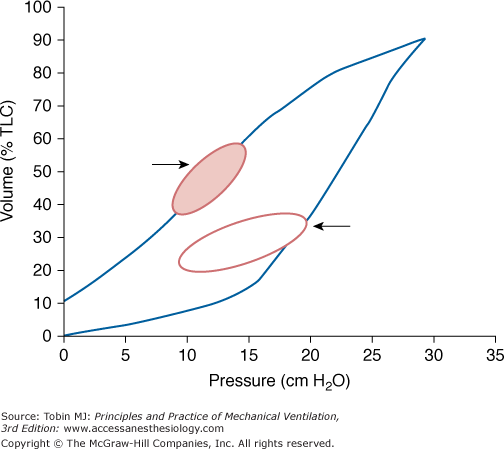
Effect on the tidal pressure volume relationship (small loops) before and after a sustained inflation recruiting maneuver. Hypothetically, first opening the lung by increasing pressure to high values allows tidal ventilation to occur at similar or lower pressures with more lung units open at end-expiration. Note that any volume gained is quickly lost after recruitment if original PEEP and position remain the same.
Because of viscoelastance and other time-dependent force-distributing phenomena, the tendency of a previously collapsed airway to open (or “yield”) is a jointly hyperbolic function of both transmural pressure and time.76 Of these, pressure is the most important variable. A variety of techniques, known as recruiting maneuvers, are designed to “open” the lung so that safe and effective combinations of PEEP and tidal volume can be utilized. Each recognizes that recruitment depends not only on the magnitude of transpulmonary pressure, but also on the duration of its application (Fig. 29-18). To consolidate benefit after a successful “recruiting maneuver,” end-expiratory pressure must remain high enough to hold open these newly recruited units once safe tidal plateau pressures are resumed. With few exceptions, this “stabilizing” value of PEEP is higher than the initial one before recruitment.77–80 Any benefit from a recruiting maneuver is extremely short lived if PEEP returns to its original value.80
Figure 29-18
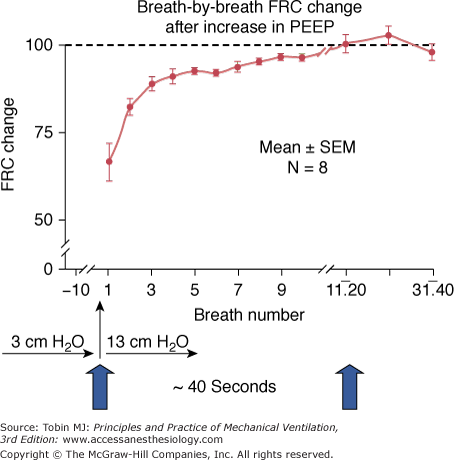
Time course of functional residual capacity (FRC
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


