Mechanical Ventilation in Chronic Obstructive Pulmonary Disease: Introduction
Acute exacerbation of chronic obstructive pulmonary disease (COPD) is defined as an increase in dyspnea, cough, or sputum production that requires therapy. The annual rate of exacerbations is 0.5 to 3.5 per patient.1 Hospitalization rates range from 0.1 to 2.4 per patient per year.1 Most exacerbations are caused by viral infections, such as Rhinovirus or influenza species or bacterial infections, such as Haemophilus influenzae, Streptococcus pneumoniae, Moraxella catarrhalis, or Pseudomonas species.2 Occasionally, exacerbations are caused by air pollution and other environmental factors.2 When evaluating a patient suspected of having an exacerbation, concurrent conditions such as pulmonary emboli, pneumothorax, and congestive heart failure should be clinically excluded.2 In about one-third of cases, no underlying etiology is identified.3
The clinical presentation of exacerbations of COPD is highly variable. Most patients require only an increase of maintenance medications, while others develop frank respiratory failure and require ventilator assistance.4,5 The goals of ventilator assistance are to decrease respiratory distress and dynamic hyperinflation, to improve gas exchange, and to buy time for resolution of the processes that triggered the episode of acute respiratory failure.
This chapter focuses on the aspects of ventilator management that are unique for patients with COPD. General principles of ventilator management are covered in other chapters—such as ventilator modes, bronchodilator therapy, weaning, and so on—and only the aspects specific to COPD are discussed in the present chapter. More than is the case with any other group of patients, clinical decision making and ventilator management in COPD is predicated on a detailed knowledge of the underlying pathophysiology.
Pathophysiologic Features Relevant to Ventilator Support
The basic physiologic abnormalities of patients who experience acute respiratory failure in COPD include deteriorations in respiratory mechanics, respiratory muscle function, and gas exchange.
In stable patients with COPD, inspiratory flow resistance is approximately 6 cm H2O/L/s above normal.6 During an episode of acute respiratory failure, resistance can increase by an additional 6 cm H2O/L/s or more.7,8 Mechanisms responsible for the increase include bronchospasm, airway inflammation, and mucus production.9 Increases in inspiratory resistance and respiratory motor output lead to increased inspiratory effort (Fig. 31-1).7 In patients with severe COPD, the pressure output of the inspiratory muscles during resting breathing is three times higher than in healthy subjects: average pressure-time products of 259 to 341 versus 94 cm H2O • second per minute.8,10 Values are five times higher when patients are in respiratory distress.8,11
Figure 31-1
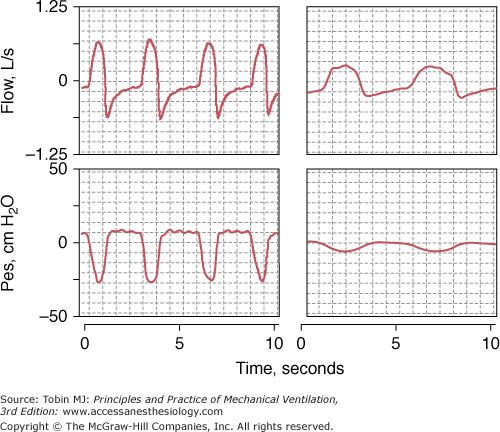
Respiratory effort during spontaneous respiration. Recordings of flow (inspiration upward) and esophageal pressure (Pes) in a patient with chronic obstructive pulmonary disease (COPD) in respiratory failure (left panel) and in a stable patient with COPD (right panel). The patient in respiratory failure exhibits a steeper fall and greater excursion in esophageal pressure than does the stable patient, signifying greater respiratory motor output. Despite the fivefold larger excursion in Pes in the patient in respiratory failure, peak inspiratory flow is only two times more than in the stable patient, signifying more abnormal mechanics in the former than in the latter patient. The patient in respiratory failure exhibits tachypnea, reflected by the shorter respiratory cycle, and exhibits a supramaximal flow transient at the beginning of exhalation, which is typical of expiratory flow limitation.
In healthy subjects, inspiratory and expiratory airway resistance are of similar magnitude.6,12 In contrast, patients with COPD have greatly increased expiratory resistance.12–14 In ventilated patients with COPD, the ratio of expiratory to inspiratory flow resistance ranges from 3.8 at low lung volumes to 1.6 at high lung volumes.14 The increased resistance at low lung volumes is related to dynamic narrowing of the small airways during exhalation.15 This narrowing is thought to result from damage to the elastic scaffold surrounding the airways16 and as well as to the “wave speed limitation” of the expiratory flow—the tracheobronchial tree cannot adjust an airflow more rapidly than the velocity at which pressure travels along the airways.15 Small-airway narrowing and wave-speed limitation reduce maximum flow during exhalation with the development of expiratory flow limitation (Fig. 31-2). Expiratory flow limitation occurs in 60% of stable patients during resting breathing,17 and universally during episodes of acute respiratory failure.18
Figure 31-2

Schematic of the isovolume pressure–flow relationship. Patients with chronic obstructive pulmonary disease (COPD) exhibit an initial diagonal segment, where increases in pressure produce increases in airflow (the effort-dependent region on the left), followed by a flat portion, where increases in pressure do not produce increases in flow (the effort-independent region on the right). Compared with a healthy subject, the slope of the initial diagonal segment is decreased, indicating an increase in airway resistance, and maximum flow is much reduced in the remaining portion secondary to expiratory flow limitation. (Used, with permission, from Tobin et al.16)
Patients with expiratory flow limitation experience air trapping whereby activation of the expiratory muscles increases alveolar pressure without decreasing the end-expiratory lung volume below the relaxation volume of the respiratory system (Vrel).15 The inability to decrease end-expiratory lung volume below Vrel has several negative consequences. First, expiratory muscle contraction—a constrained response of the respiratory centers to increased ventilatory demands—does not achieve storage of elastic energy in the respiratory system at end exhalation.4 Second, relaxation of the expiratory muscles at the onset of inhalation cannot assist the inspiratory muscles in expanding the respiratory system during inhalation. Third, the entire burden of breathing is borne by the inspiratory muscles. Fourth, recruitment of the expiratory muscles dissipates precious energy substrates. That is, expiratory muscle recruitment in COPD is harmful,19 and it can account for 66% of the variation in Borg scale ratings of difficulty in breathing.20
The minimum time required to exhale from a given lung volume to Vrel is determined by the maximum expiratory flow.15 When the time for exhalation (TE) is less than this minimum time, inhalation will begin before the respiratory system has returned to Vrel—a state known as dynamic hyperinflation (Fig. 31-3).16 Dynamic hyperinflation (a “volume” phenomenon) is almost invariably associated with an increase in end-expiratory elastic recoil of the respiratory system (a “pressure” phenomenon).15 This increase in end-expiratory elastic recoil has been called auto–positive end-expiratory pressure (auto-PEEP)21 or intrinsic PEEP (PEEPi).22 Dynamic hyperinflation and PEEPi have been reported in all ventilated patients with COPD.23 Increases in dynamic hyperinflation during exacerbations are proportionally greater than increases in airflow obstruction.24
Figure 31-3
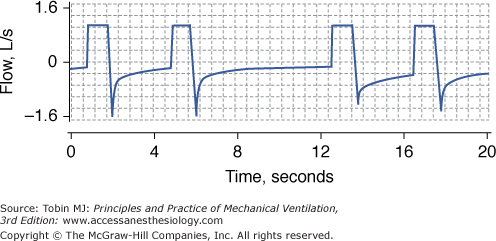
Persistent expiratory flow at end-exhalation. Recording of flow (inspiration upward) in a patient with chronic obstructive pulmonary disease (COPD) and respiratory failure receiving controlled mechanical ventilation. Whether the duration of exhalation is 3 seconds (first and third breaths) or 6 seconds (second breath), expiratory flow at end-exhalation is always present. Persistent expiratory flow at end-exhalation signifies that inhalation begins before the respiratory system has returned to its relaxation volume—dynamic hyperinflation.
Dynamic hyperinflation and PEEPi can fluctuate widely as a result of several factors.15 First, bronchospasm, mucosal edema, and sputum inspissation can abruptly worsen expiratory flow limitation. Second, patients are often tachypneic and, thus, TE is shortened.25 Finally, ventilatory demands can fluctuate because of sudden episodes of anxiety or deterioration of gas exchange.15
Acute worsening in dynamic hyperinflation is beneficial because it optimizes expiratory flow by limiting dynamic airway narrowing during exhalation26 and by increasing the elastic recoil of the lung and, less so, of the chest wall.18 It is detrimental because it decreases the effectiveness of the respiratory system through several mechanisms. First, at the start of inhalation patients have to first generate negative inspiratory pressure equal in magnitude to the value of PEEPi before inspiratory flow can be initiated (threshold inspiratory load).8,27 In patients with COPD recovering from an episode of respiratory failure necessitating mechanical ventilation, Jubran and Tobin8 reported that, during a spontaneous breathing trial, approximately 20% to 25% of the inspiratory muscle effort was required to overcome PEEPi. Second, dynamic hyperinflation impairs respiratory muscle function by worsening the length–tension relationship of the muscles (Fig. 31-4), by decreasing the zone of apposition,28 and by interfering with inspiratory muscle perfusion.29 Finally, dynamic hyperinflation increases end-inspiratory lung volume.18 When excessive, increases in end-inspiratory lung volume can reduce lung compliance, increase the elastic work of breathing, and can cause alveolar overdistension. Alveolar overdistension can lead to hemodynamic compromise through several mechanisms. First, it can reduce right-ventricular preload as a result of impaired venous return.21 Second, it can raise right-ventricular afterload by increasing pulmonary vascular resistance.21 The increased right-ventricular afterload can increase the right ventricular end-diastolic volume and shift the interventricular septum toward the left ventricle. This shift can impair left-ventricular filling (ventricular interdependence). Finally, the threshold load imposed on the inspiratory muscles can increase left-ventricular afterload secondary to increased negative intrapleural pressure during inhalation.21 All these phenomena can severely decrease cardiac output, effects exaggerated in COPD because the abnormally compliant lungs transmit a high fraction of alveolar pressure to intrathoracic vessels.30
Figure 31-4
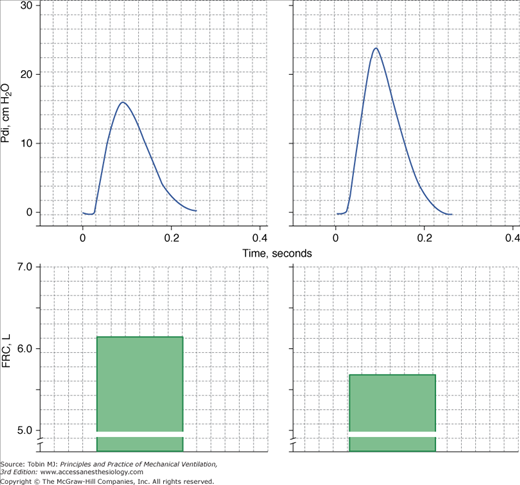
Twitch transdiaphragmatic pressure (Pdi) elicited by phrenic nerve stimulation (upper panels) and functional residual capacity (FRC; lower panels) in a patient with chronic obstructive pulmonary disease (COPD) before (left) and after (right) lung volume reduction surgery. The higher twitch Pdi after surgery was in part caused by decrease in operating lung volume as demonstrated by decrease in FRC. (Data from Laghi et al.10)
Patients with COPD do not generate as much negative maximal inspiratory pressures as do healthy subjects.4 This decrease in respiratory muscle strength may result from several factors, including greater protein degradation of muscle fibers,31,32 malnutrition, sepsis, systemic steroids, and ventilator mode.4,28 In some patients, however, the inspiratory weakness can be completely explained by hyperinflation-induced muscle shortening.33 In addition, diaphragmatic myofibers of patients with COPD are also more susceptible to sarcomere disruption when subjected to an acute inspiratory load than are the diaphragms of healthy controls.34
Patients with COPD have an increased risk of coronary artery disease, and 20% to 30% have chronic heart failure.35 Increased stress on the myocardium during acute respiratory failure could overwhelm an already impaired cardiac reserve.36,37 An increased load resulting from interstitial edema secondary to acute left-ventricular failure together with decreased blood flow to the respiratory muscles may markedly impair respiratory muscle performance.38
Contractile fatigue occurs when a sufficiently large respiratory load is applied over a sufficiently long period of time. Contractile fatigue can be brief or prolonged.28 Short-lasting fatigue results from accumulation of inorganic phosphate, failure of the membrane electrical potential to propagate beyond T-tubules, and to a much lesser extent intramuscular acidosis.28 In nine patients who developed acute respiratory failure during a weaning trial (four of whom had COPD), Brochard et al39 reported electromyographic signs suggestive of incipient short-lasting diaphragmatic fatigue. Short-lasting fatigue appears to have a protective function, because it can prevent injury to the sarcolemma caused by forceful muscle contractions.40 Long-lasting fatigue41 is consistent with the development of, and recovery from, muscle injury.28,40
To assess whether patients in acute respiratory failure develop long-lasting fatigue, Laghi et al11 measured the contractile response of the diaphragm to phrenic nerve stimulation in sixteen patients being weaned from mechanical ventilation. In that study, no patient developed long-lasting fatigue of the diaphragm. The investigators concluded that patients displayed clinical manifestations of severe respiratory distress for a substantial time before they would develop fatigue.
Abnormal distribution of ventilation–perfusion ( ) ratios42 and decreased mixed venous oxygen tension are common causes of hypoxemia during exacerbations.43,44 In patients without alveolar pathologies (pneumonia, pulmonary edema) shunt is almost negligible.43
) ratios42 and decreased mixed venous oxygen tension are common causes of hypoxemia during exacerbations.43,44 In patients without alveolar pathologies (pneumonia, pulmonary edema) shunt is almost negligible.43
Hypercapnia is present in nearly half of patients admitted to a hospital with an acute exacerbation of COPD.45 Hypercapnia plus respiratory acidosis is almost universal in patients with COPD requiring mechanical ventilation.45
Excluding patients who are on the verge of respiratory arrest, most patients with COPD in respiratory failure who develop hypercapnia experience a decrease in alveolar ventilation ( ) as a result of a change in the pattern of breathing and not as a result of decreased respiratory drive.25 The dominant finding of this pattern of breathing is a shortening of inspiratory time (TI) and TE.25 These combined changes lead to a marked increase in respiratory frequency and decrease in tidal volume (VT)—rapid shallow breathing. The decrease in VT results in increased dead-space-to-tidal-volume ratio (VD/VT) or dead space ventilation that is not compensated by the increase in minute ventilation. In a study of seventeen patients with COPD requiring mechanical ventilation, Tobin et al25 reported that the combined changes in VT and respiratory frequency accounted for 81% of increase in partial pressure of arterial carbon dioxide (PaCO2) in the ten patients who developed respiratory distress when disconnected from mechanical ventilation. Whether rapid shallow breathing is an adaptive response to the mechanical constraints of acute dynamic hyperinflation that minimizes dyspnea and lessens the risk of respiratory muscle fatigue remains to be determined.
) as a result of a change in the pattern of breathing and not as a result of decreased respiratory drive.25 The dominant finding of this pattern of breathing is a shortening of inspiratory time (TI) and TE.25 These combined changes lead to a marked increase in respiratory frequency and decrease in tidal volume (VT)—rapid shallow breathing. The decrease in VT results in increased dead-space-to-tidal-volume ratio (VD/VT) or dead space ventilation that is not compensated by the increase in minute ventilation. In a study of seventeen patients with COPD requiring mechanical ventilation, Tobin et al25 reported that the combined changes in VT and respiratory frequency accounted for 81% of increase in partial pressure of arterial carbon dioxide (PaCO2) in the ten patients who developed respiratory distress when disconnected from mechanical ventilation. Whether rapid shallow breathing is an adaptive response to the mechanical constraints of acute dynamic hyperinflation that minimizes dyspnea and lessens the risk of respiratory muscle fatigue remains to be determined.
In some patients with COPD, hypercapnia can be worsened by the administration of supplemental oxygen.45 This risk is significant when the inspired oxygen concentration exceeds 30%. Mechanisms that may contribute to CO2 retention include a decrease in hypoxic ventilatory response consequent to the administration of oxygen; an increase in dead space consequent to release of hypoxic vasoconstriction and, thus, worsening of 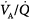 relationships; and the Haldane effect (for any given amount of CO2 bound to hemoglobin PaCO2, is considerably higher in the presence of a high versus a low oxygen saturation).16
relationships; and the Haldane effect (for any given amount of CO2 bound to hemoglobin PaCO2, is considerably higher in the presence of a high versus a low oxygen saturation).16
Ventilator Assistance
The many pathophysiologic defects considered above have direct bearing to the judicious administration of mechanical assistance in patients with COPD in respiratory failure. Physicians considering the need to implement mechanical ventilation in these patients have to answer five basic questions:
When should mechanical ventilation be started, and which goals should be pursued?
How should mechanical ventilation be delivered—noninvasive versus invasive?
What setting and mode of mechanical ventilation should be used?
When—and how—should mechanical ventilation be discontinued?
What can be done when weaning is difficult or impossible?
Most patients with mild exacerbations of COPD (70% to 90% of those hospitalized for an exacerbations)5,45,46 can be cared for with oxygen and pharmacotherapy alone. The remaining 10% to 30% require ventilator assistance.5,45–47
Noninvasive positive-pressure ventilation (NIPPV) has become the first line intervention for most patients with COPD in acute respiratory failure (Table 31-1).5 NIPPV decreases the need for endotracheal intubation and the rate of ventilator-related complications, intensive care unit (ICU) admissions, and in-hospital and 1-year mortality.47–49
|
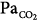
In most studies, the rate of failure with NIPPV is inversely related to the severity of respiratory acidosis. When pH is 7.30 to 7.34 on admission, the rate of NIPPV failure is 10% to 20%;47,50 when pH is 7.25 to 7.30, NIPPV failure is 30% to 40%;51,52 and when pH is less than 7.25, failure is 50% to 60%.53,54 These rates, however, have to be interpreted with caution. In some series, NIPPV failure in patients with very low pH (7.13 to 7.20) on admission was only 14% to 35%.55–57
Failure with NIPPV usually occurs in the first hours of ventilator support.58 Mask intolerance, uncontrollable leaks, and lack of improvement in gas exchange are the most common causes of failure.59,60 Early improvement should not lull the physician into a false sense of security: Approximately 20% of initial responders (first 48 hours) experience a second episode of acute respiratory failure.60 Late-onset deterioration is more likely in patients with worse clinical condition before admission, and more complications at admission.60 Patients who experience late failure have a poor inhospital prognosis, particularly if they are not promptly intubated.60
Negative pressure ventilation (iron lung ventilation) has been successfully used in patients with COPD and severe respiratory acidosis (pH < 7.25).61–63 (See Chapter 16.)
NIPPV and negative pressure ventilation do not protect the upper airway in patients who are gasping, have respiratory arrest or have lost consciousness.47 NIPPV and negative pressure ventilation are also unsafe and poorly tolerated in patients requiring high levels of inspiratory airway pressure (>25 to 30 cm H2O) and inspired oxygen (FIO2 > 0.60). These patients, as well as those who are hemodynamicaly unstable despite fluid resuscitation and vasopressors, and those who are agitated, uncooperative, or who fail noninvasive ventilation, require endotracheal intubation and invasive ventilation (see Table 31-1). Despite earlier concerns, severe hypercapnic encephalopathy (Glasgow Coma Scale score ≤8 to 10) should not—a priori—be considered an absolute contraindication to NIPPV.55,57,64,65 If NIPPV is used in encephalopathic patients, opinions vary as to the advisability of inserting a nasogastric tube to decrease the risk of aspiration of gastric contents.55,57,64
The primary goals of ventilator assistance are to decrease inspiratory effort and respiratory distress, to minimize dynamic hyperinflation and PEEPi, to lessen respiratory acidosis, and to improve hypoxemia.
Most patients with COPD in respiratory failure are dyspneic and have clinical signs of increased work of breathing: nasal flaring, vigorous activity of the sternomastoid muscles, tracheal tug, recession of the suprasternal, supraclavicular and intercostal spaces, paradoxical motion of the abdomen, and pulsus paradoxus.66 In these patients, invasive67,68 and noninvasive69–71 ventilation can decrease inspiratory effort and respiratory distress (Fig. 31-5).
Figure 31-5

Inspiratory effort, quantitated as by esophageal pressure-time product per minute (PTP/min), and dyspnea, quantitated by Borg score in eleven ventilator-dependent patients with chronic obstructive pulmonary disease (COPD) while receiving pressure-support ventilation (PSV) and intermittent mandatory ventilation (IMV). Left panel: PTP decreased as the level of PSV or IMV was increased. At proportional levels of ventilatory assistance, PTP/min was not different during PSV and IMV (p < 0.0005 in both instances). Right panel: Dyspnea decreased with increasing levels of PSV or IMV (p < 0.05 in both instances). Bars represent ± standard error (SE). (Modified, with permission, from Leung et al.67)
Mechanical ventilation can both worsen or improve dynamic hyperinflation and PEEPi.15 Dynamic hyperinflation and PEEPi should be suspected in any patient with persistent expiratory flow (see Fig. 31-3) at end-exhalation.15 The magnitude of end-expiratory flow, however, bears little relation to the magnitude of PEEPi.30
Mechanical ventilation improves hypercapnia by decreasing CO2 production and, more importantly, by increasing 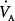 . Vigorous increases in minute ventilation should be avoided, because of risk of iatrogenic dynamic hyperinflation. When dynamic hyperinflation is a concern, and provided that intracranial hypertension and overt hemodynamic instability do not exist, acceptance of acidemia (pH > 7.2) may be reasonable.72
. Vigorous increases in minute ventilation should be avoided, because of risk of iatrogenic dynamic hyperinflation. When dynamic hyperinflation is a concern, and provided that intracranial hypertension and overt hemodynamic instability do not exist, acceptance of acidemia (pH > 7.2) may be reasonable.72
In addition to worsening dynamic hyperinflation, overzealous ventilation can cause life-threatening alkalosis.66 Severe alkalosis can cause coronary artery spasm, central nervous system hypoperfusion, myoclonus, asterixis, and seizures. Other effects of alkalosis include increase in hemoglobin affinity for oxygen, and, in the presence of increased shunt, a possible worsening of the  relationship (secondary to a decrease in hypoxic pulmonary vasoconstriction).66 Excessive ventilation, over time, causes bicarbonate wasting by the kidney. In patients who retain CO2 when clinically stable, this renal wasting of bicarbonate will increase ventilatory demands during weaning.
relationship (secondary to a decrease in hypoxic pulmonary vasoconstriction).66 Excessive ventilation, over time, causes bicarbonate wasting by the kidney. In patients who retain CO2 when clinically stable, this renal wasting of bicarbonate will increase ventilatory demands during weaning.
A wide variety of ventilator modes are used in COPD. These include pressure-support ventilation (PSV), assist-control ventilation (ACV) that can be either volume-cycled or pressure-cycled (PCV), intermittent mandatory ventilation, and, more recently, proportional-assist ventilation and neurally adjusted ventilatory assist. Each of these modes is covered in detail in other chapters.
It is unknown whether one ventilator mode is superior to another in patients with COPD. I summarize here my own preferences, and the physiologic rationale behind my choices. In patients requiring NIPPV, I usually use PSV5,73 based on two considerations. First, unlike volume-cycled modes (ACV, intermittent mandatory ventilation), PSV allows breath-by-breath titration of support. The hope is to improve patient comfort and thus achieve greater success with NIPPV. Second, PSV is equally as effective as volume-cycled modes in decreasing inspiratory effort (see Fig. 31-5). I add a moderate amount of external PEEP to counterbalance PEEPi—a combination that lowers inspiratory work more than the use of PSV or PEEP alone.74,75 Persistent respiratory distress despite high levels of support can result from expiratory muscle recruitment.76 This recruitment can be caused by inadequate “cycling-off” criteria of PSV: In patients with COPD the decay in inspiratory flow during PSV is less steep than in patients with normal respiratory mechanics.76,77 When expiratory muscle recruitment does occur an increase in the threshold of peak inspiratory flow to cycle off PSV can improve patient–ventilator interaction.77,78
Persistent respiratory distress despite high levels of support can result from excessive load on the respiratory muscles. In this situation, an increase in inspiratory pressurization rate—the time needed to reach the target inspiratory pressure—can decrease inspiratory effort.79 Unfortunately, increases in pressurization rate during NIPPV are associated with increased air leaks and decreases a patient’s tolerance of NIPPV.79 When respiratory distress persists despite high levels of support, it may be more prudent to intubate a patient.
In intubated patients, I mostly use ACV80 and, occasionally, PCV. I prefer ACV and PCV over PSV in intubated patients because these patients commonly receive sedation. Sedation can decrease respiratory motor output and thus promote alveolar hypoventilation. When using ACV I usually set the inspiratory flow waveform in the square pattern to facilitate monitoring of mechanics. When intubated patients with COPD display a high drive, I may use PCV. With this mode, patients have more control over the peak flow than with ACV. I avoid controlled mechanical ventilation because it is associated with the early development of respiratory muscle atrophy and damage.81,82
Proportional-assist ventilation and neurally adjusted ventilatory assist are two novel modes, but it is not known whether they will prove superior to traditional ventilator modes.
The most valuable approach to reduce or eliminate PEEPi is to diminish minute ventilation,30 even if this means development of alveolar hypoventilation—permissive hypercapnia.83 In patients with PEEPi ventilated with volume-cycled modes, clinicians often increase inspiratory flow to decrease TI. This is done hoping to prolong TE. Increases in inspiratory flow, however, commonly lead to an increase in respiratory rate84,85 and the anticipated reduction in TE could increase PEEPi. Laghi et al85 studied this phenomenon in ten patients with COPD while they were receiving ACV with a backup rate of 1 breath/min. As expected, an increase in flow from 30 to 90 L/min increased the respiratory rate from 16 ± 1 to 21 ± 2 breaths/min. Despite the rise in rate, PEEPi decreased from 7 ± 1 to 6 ± 1 cm H2O. The decrease in PEEPi was the result of a paradoxical increase in TE, which permitted extra time for lung deflation. The investigators reasoned that the shortened TI secondary to the increase inspiratory flow combined with time-constant inhomogeneity of COPD caused overinflation of some lung units to persist into neural exhalation. Continued inflation during neural exhalation could stimulate the vagus, which prolongs expiratory time.84,85
In patients with COPD and flow-limited physiology, low levels of external PEEP can maintain a patent (or nearly patent) channel between the central airway and the alveolar compartment at the start of inhalation. In these patients, application of low-level external PEEP reduces the effort to trigger the ventilator,14
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


