CHAPTER 7
Local Anesthetics
INTRODUCTION
Local anesthetics (LAs) are used in many clinical scenarios that require the pain practitioner to be intimately familiar with their pharmacokinetic and pharmacodynamic properties. They are used in the prevention of acute, inflammatory, nociceptive, cancer-related, neuropathic, and chronic pain, for diagnostic and prognostic purposes, and in the possible prevention of chronic pain.1
Operating primarily by their interaction with sodium voltage dependent channels (NaVs) to block sodium (Na+) entry into the neuron, LAs interrupt nerve impulse conduction and propagation. However, in recent years, other actions, such as LA interaction with other non-Na+ ion channels and G-protein receptors, which may be important in their ability to prevent and treat pain, have been described.2 These interactions take place in both the central and peripheral nervous system. In a concentration dependent fashion, LAs interrupt autonomic, sensory, and motor impulses, giving rise to autonomic system blockage, sensory analgesia, and muscle paralysis. With a deeper understanding of both the pharmacokinetics and pharmacodynamics of LAs, a higher rate of treatment success can be achieved, with a reduction in side effects. This chapter aims to focus on the newest information regarding LA mechanisms, pharmacodynamics, and toxicity.
PREPARATIONS
LAs, being poorly soluble in water, are mixed with hydrochloride salts. If epinephrine is added to the solution, sodium bisulfate is often added to further lower the pH to about 4 to prevent its oxidative decomposition.
LAs such as lidocaine, tetracaine, and bupivacaine are also manufactured in liposomal vesicles. Such a formulation provides for a prolongation of action and a decrease in toxicity.3 Such a formulation allows for the slow continuous subcutaneous infiltration of the drug providing analgesia for up to 96 hours to decrease toxicity.4
Since the pKa of most LAs is approximately 8, within the acidic carrier medium they are dissolved in, pH 3.9 to 6.5, only approximately 3% of the drug exists in the lipid-soluble form. Because the lipid-soluble form is felt important for LA mechanism of action, alkalinization, via addition of sodium bicarbonate, is used by some practitioners. This process shifts the pH of the medium to become closer to the pKa of the drug, allowing for more of the drug to exist in the lipid-soluble form. The result is a hastening of the onset of LAs in peripheral nerve and neuraxial blocks by 3 to 5 minutes, a deepening of the depth of sensory and motor block, and an increase in the epidural spread of the drug.5 However, prolonged or excessive alkalinization can cause LA molecules to precipitate from suspension. Additionally, the value of adding sodium bicarbonate to solutions to enhance speed of onset also depends on the injection site and the specific physiochemical properties of the individual LA used.6
STRUCTURE
LAs consist of 4 portions:
1. Aromatic ring
2. Ester or amide linkage
3. Hydrocarbon chain
4. Tertiary amine group (Figure 7-1)
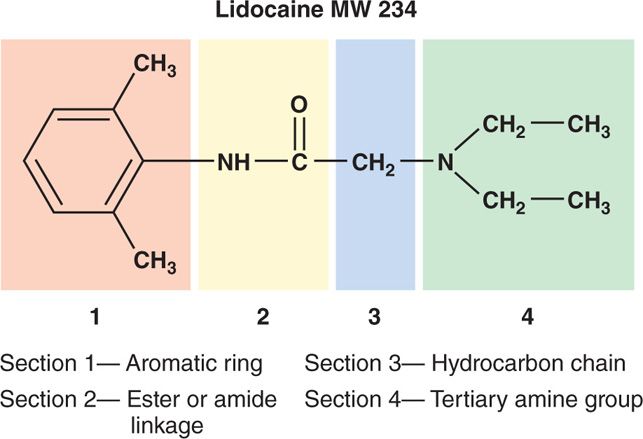
Figure 7-1. The four parts of a local anesthetic molecule. Local anesthetics consist of four portions: (1) aromatic ring, (2) ester or amide linkage, (3) hydrocarbon chain, and (4) tertiary amine group.
By analyzing the type of bond that conjoins the hydrocarbon chain to the lipophilic aromatic ring, two types of LA classes are discerned; the amino ester–linked local anesthetics and the amino amide–linked local anesthetics (Figure 7-2).
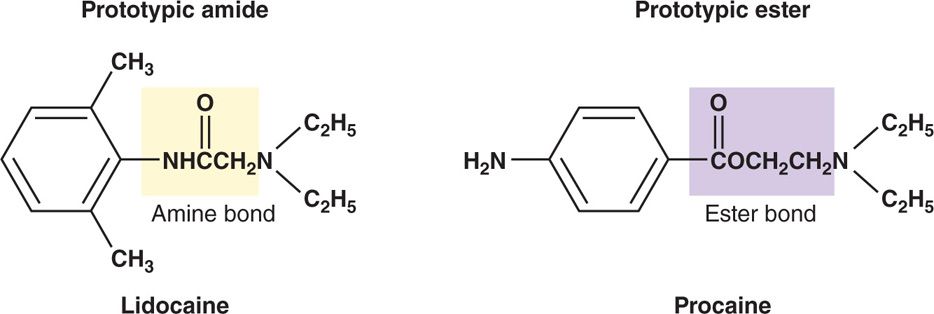
Figure 7-2. Prototypic amino amide, lidocaine, and amino ester, procaine, local anesthetics. Note the amide bond conjoining the hydrocarbon chain of the lidocaine molecule to the aromatic ring, and the ester bond conjoining the hydrocarbon chain of the procaine molecule to the aromatic ring. These different types of bonds are what distinguish the two classes of local anesthetics.
Four LAs are characterized as pipecoloxylidides:
1. Mepivacaine
2. Bupivacaine
3. Levobupivacaine
4. Ropivacaine
Because the pipecoloxylidides have an asymmetric carbon atom, they are chiral drugs, which means they can have a left (S) or right (R) handed configuration. These configurations are mirror images of each other and are known as enantiomers or stereoisomers (Figure 7-3).
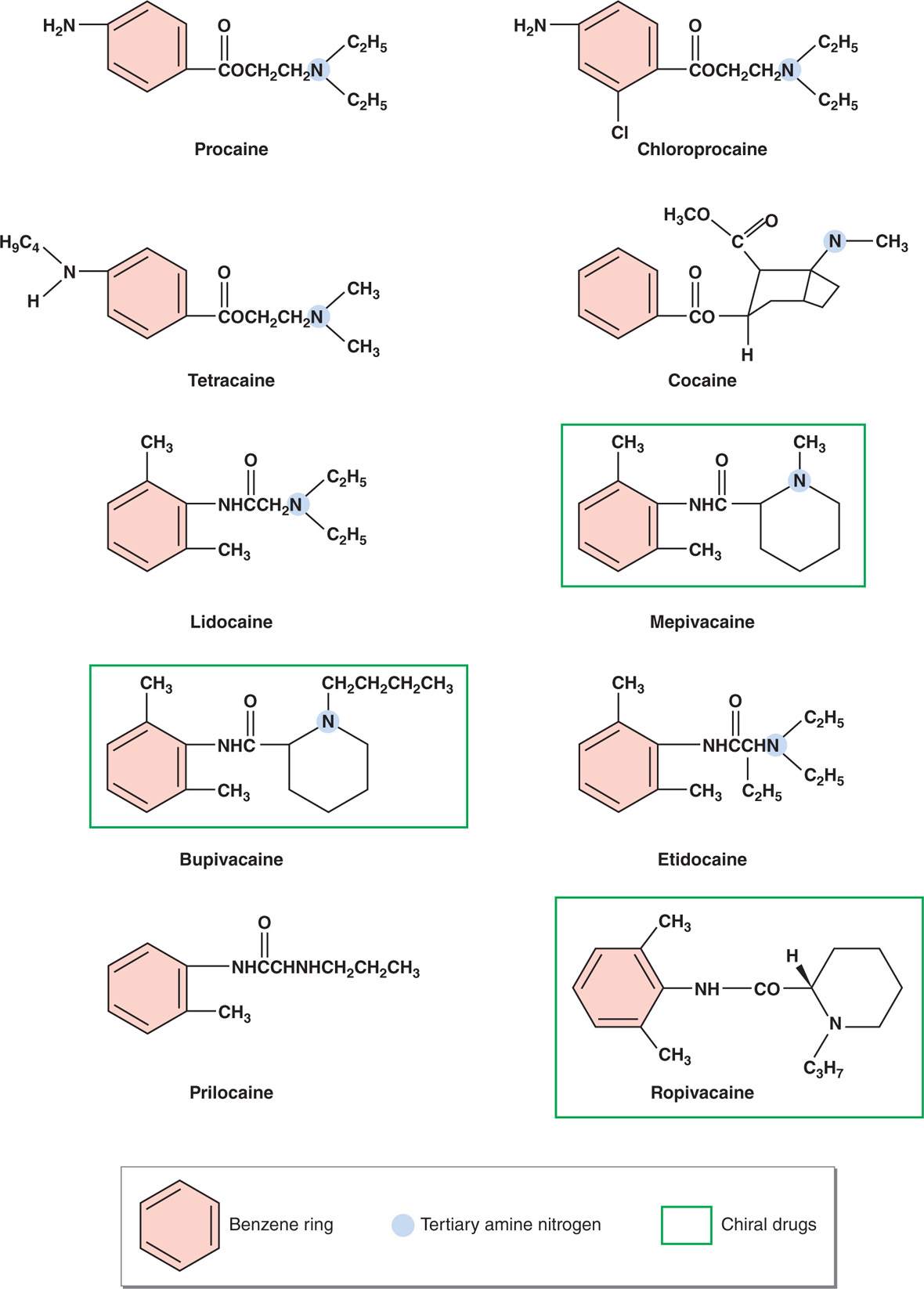
Figure 7-3. Chemical structure of ester and amide local anesthetics. Esters and amides share many features, including a substituted benzene ring at one end of the molecule, and a tertiary amine nitrogen at the other end. Note that prilocaine is an exception in that it has a secondary amine on its hydrophilic end. Mepivacaine, bupivacaine, and ropivacaine are chiral drugs because the molecules possess an asymmetric carbon. The red underscore line highlights the common benzene ring, the blue arrow highlights the common tertiary amine nitrogen, the green underscore highlights the chiral carbon, and the green box highlights the chiral drugs.
Aside from ropivacaine and levobupivacaine, all other LAs are either racemic mixtures, or have no asymmetric (chiral) carbons.
It is easy to appreciate that these two forms of chiral LAs, the S and R forms, have their own pharmacodynamic, pharmacokinetic, and toxicity profiles.7 Generally speaking in reference to LAs, the S form is less toxic, more potent, and longer acting than the R form or the racemic mixture.6
Because of these observations, ropivacaine and levobupivacaine were developed as pure S enantiomers, which offer less neurotoxicity and cardiotoxicity.8
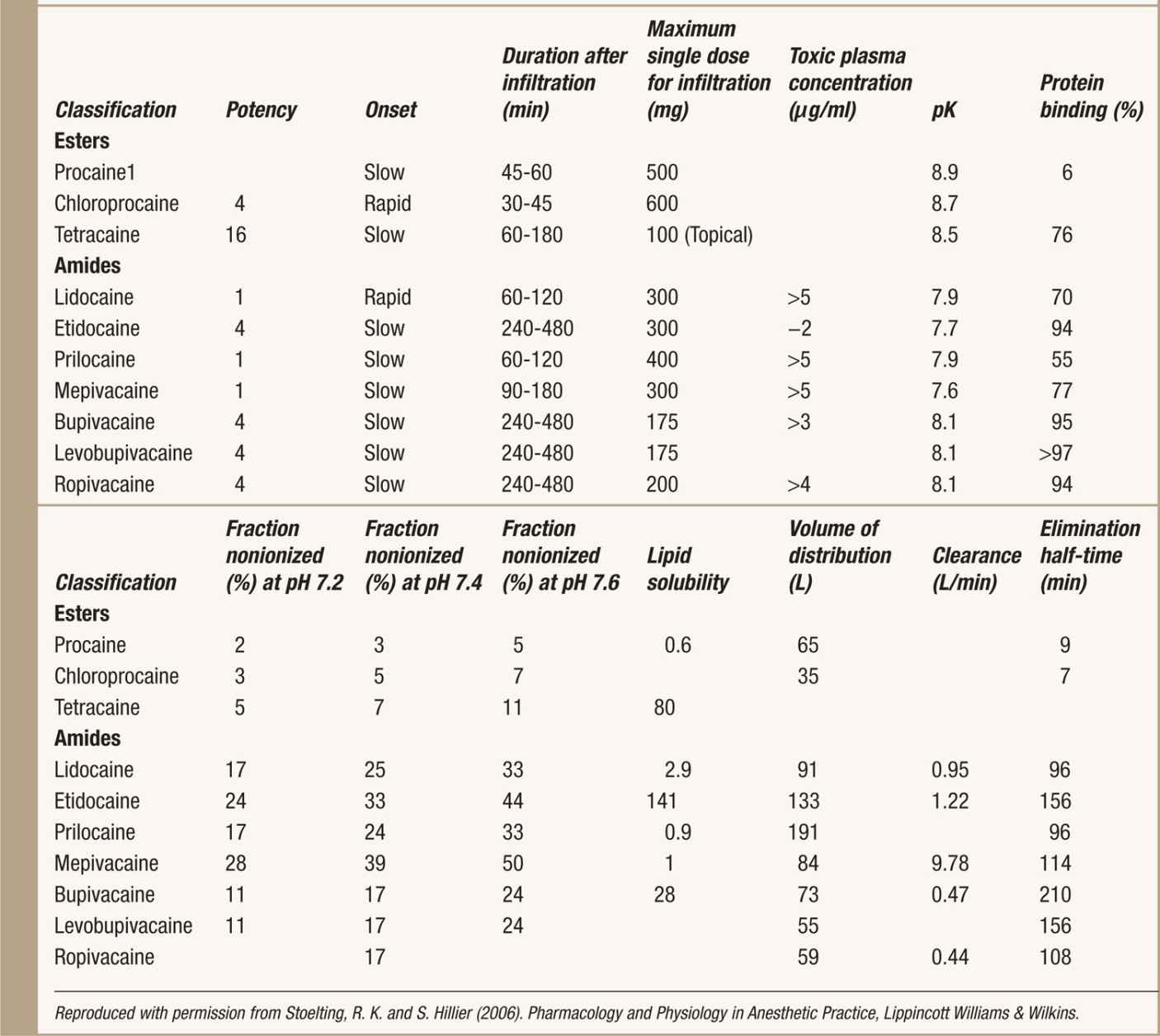
A complete comparative pharmacology of LAs can be found in Table 7-1.
TABLE 7-1. Comparative Pharmacology of Local Anesthetics
As evident in Figure 7-4, since the creation of chloroprocaine in 1955, emphasis has shifted toward the development of amide LAs.
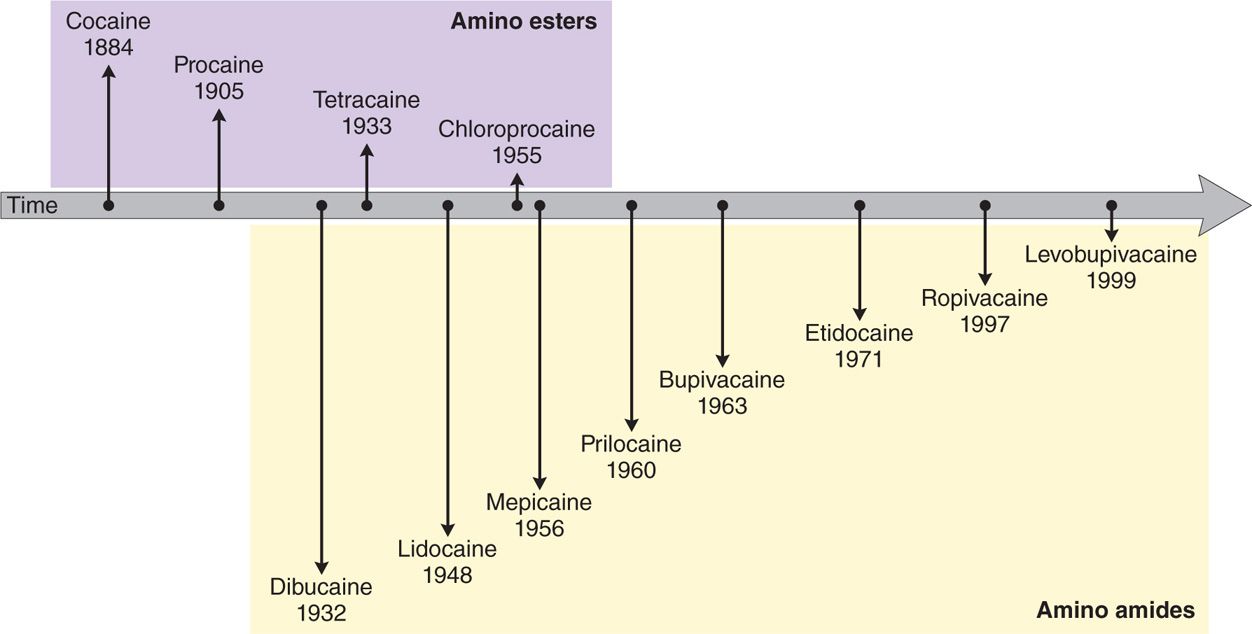
Figure 7-4. Chronology of the development of local anesthetics.
This is due to the allergenic potential of esters, as well as their instability. As the evolution of amide LAs progressed, the testing of various modifications to the template structures of procaine and lidocaine revealed that increasing the molecular weight of the molecules, by adding carbon atoms to either end of the structure or to the amide link, generally increases the lipid solubility, protein binding, duration of action and toxicity, and influences biotransformation and metabolism of the molecule.6
MECHANISM OF ACTION
Voltage-gated sodium channels, NaV1.1–NaV1.9, are essential for voltage sensing, generation, and ion conduction of electrical impulses in excitable cells.9 Na+ ions are naturally permeable through these ion-selective channels. Simplistically speaking, by inhibiting the passage of Na+ ions through these ion-selective sodium channels, by preventing them from “opening,” LAs prevent threshold potential, and, hence, action potential propagation10 (Figure 7-5).
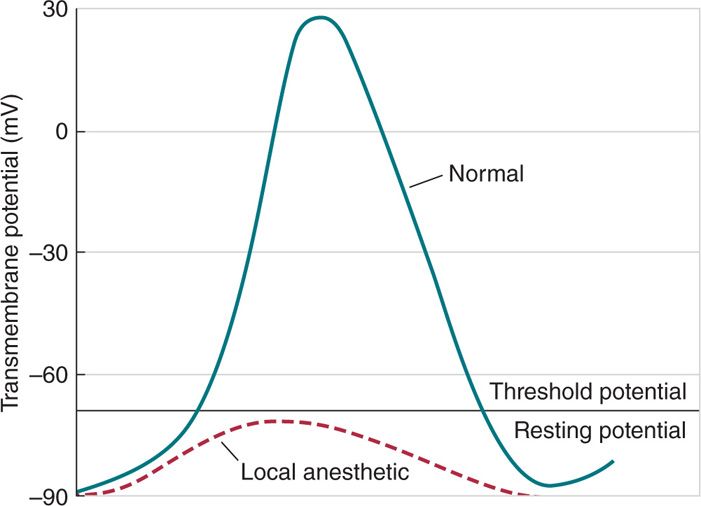
Figure 7-5. How local anesthetics interact with sodium channels to prevent threshold activation Local anesthetics slow the rate of depolarization of the nerve action potential such that the threshold potential is not reached. As a result, an action potential cannot be propagated in the presence of local anesthetic, and conduction blockade results.
NaV channels are divided into those that are tetrodotoxin sensitive (TTX-S Na+) and tetrodotoxin resistant (TTX-R Na+) channels.
• Sensory neurons tend to express tetrodotoxin sensitive NaV channels.
• In contrast, many nociceptive A-delta and C-fibers expressed in dorsal root ganglion cells express tetrodotoxin-resistant NaV channels. There is evidence that channels from both groups are involved in pain states.
If used in lower concentrations, LAs can produce sensory block with little or no motor block.
A number of factors contribute to sensory versus motor differential block:
• Including anatomic factors, the relative binding affinity of different local anesthetics to the variety of NaV channels
• As well as possible contribution from other ion channels such as potassium (K) channels6
The administration of systemically administered intravenous lidocaine produces analgesia in several acute and chronic conditions.9,11–18 The antinociceptive effect of bupivacaine may be due to direct modulation of NMDA receptors in the superficial dorsal horn.19 Cited most commonly is LA binding to normal or altered NaV channels located throughout the pain processing pathways of the body as the main site of action for pain sensation modulation. It has been shown that lidocaine infusions (most commonly 5 mg/kg IV) or mexiletine are as effective as morphine, gabapentin, amitriptyline, and amantadine in the treatment of neuropathic pain.20
Many chronic pain conditions, including complex regional pain syndromes I and II, are exacerbated by sympathetic fibers that sprout into the dorsal root ganglia (DRG) after peripheral nerve injury, forming abnormal connections with sensory neurons.21 Systemic lidocaine administered at the time of surgery via an implanted osmotic pump remarkably reduces sympathetic sprouting two- to threefold.18 These effects of systemic lidocaine lasted more than 7 days after the termination of administration.18 Nerve growth factor (NGF) also contributes to the sympathetic sprouting. Lidocaine, bupivacaine, and procaine have been shown to decrease NGF signaling by suppression of NGF-stimulated neurite outgrowth in PC12 cells, which is a cellular model of sympathetic sprouting.22
VOLTAGE-GATED NA (NaV) CHANNELS
NaV channels exist in 3 states:
• Resting-closed
• Activated-open
• Inactivated-closed
These correspond to the nerve membrane cycle of polarized, depolarized, and repolarized during action potential evolution (Figure 7-6).
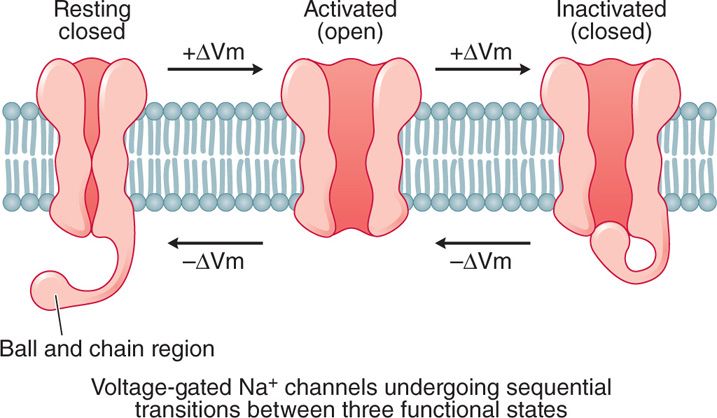
Figure 7-6. Functional states of voltage-gated sodium channels. Voltage-gated sodium channels exist in three states, resting-closed, activated-open, and inactivated-closed, corresponding to the nerve membrane cycle of polarized, depolarized, and repolarized during action potential evolution.
The resting-closed NaV channel represents the resting polarized state of the nerve fiber, the activated-open state allows Na+ ions to flow into the cell.
• While the inactivated-closed state is impermeable to Na+ ions; hence Na+ flux ceases during this phase, which represents the refractory period, and is an important element for the propagation of action potentials down an axon.10
The affinity of LAs binding to NaV channels is ordered according to the conformational state of the NaV channel, with the arrangement being activated-open > inactivated-closed > resting-closed.6 In addition to the conformational state, stereospecificity, of both the LA and NaV channel is also important. LAs bind primarily to the activated-open state, stabilizing this conformation and preventing the eventual change of the channel to the resting-closed state in response to action potential propagation.
Upon closer examination, channels in the inactivated state are believed to be blocked by LAs on their intracellular side at a specific site, a so called inner gate, activation gate, or H gate. This notion is also consistent with LA potency being related to its lipid-soluble state allowing passage into the cell. This gate is then removed in response to stimulation that opens the channel.
Once inside the cell, the low pH causes a greater fraction of the LA to assume an ionized state, and it is this ionized state that binds to receptor.10 Upon LA binding to the intracellular portion of the receptor, it is thought that a tethered plug is formed to produce an inactivation gate.
LAs also appear able to bind to the external opening of the NaV channel in the inactivated-closed state. Because of their different chemical structures, LAs bind to differing sites on NaV channels along a continuum, and binding is weak secondary to a poor fit. This poor binding helps to explain why the inclusion of epinephrine to most LA helps prolong the binding opportunities of the LA to the receptor. By decreasing blood flow to the receptor, epinephrine can help decrease the concentration gradient, between the NaV channel and the regional blood vessels, across which the LA can diffuse.
Though the detailed description of the NaV channel above centered around LAs, it is important to note that many compounds can inhibit these channels other than LAs, including general anesthetics, α2-agonists, tricyclic antidepressants, alcohols, and toxins.10,23–28
SODIUM VOLTAGE DEPENDENT CHANNELS IN PAIN STATES
The responses of nociceptors to stimuli are encoded by action potentials, whose genesis and propagation are dependent on NaVs, and it is thus not surprising that aberrant expression patterns of channels and inherited sodium channelopathies have been linked to neuropathic and inflammatory pain.9
• Humans have a total of 9 functional genes that code for NaV channels.
• Seven of these genes code for neuronal NaV channels, and 2 codes for skeletal or cardiac NaV channels.16
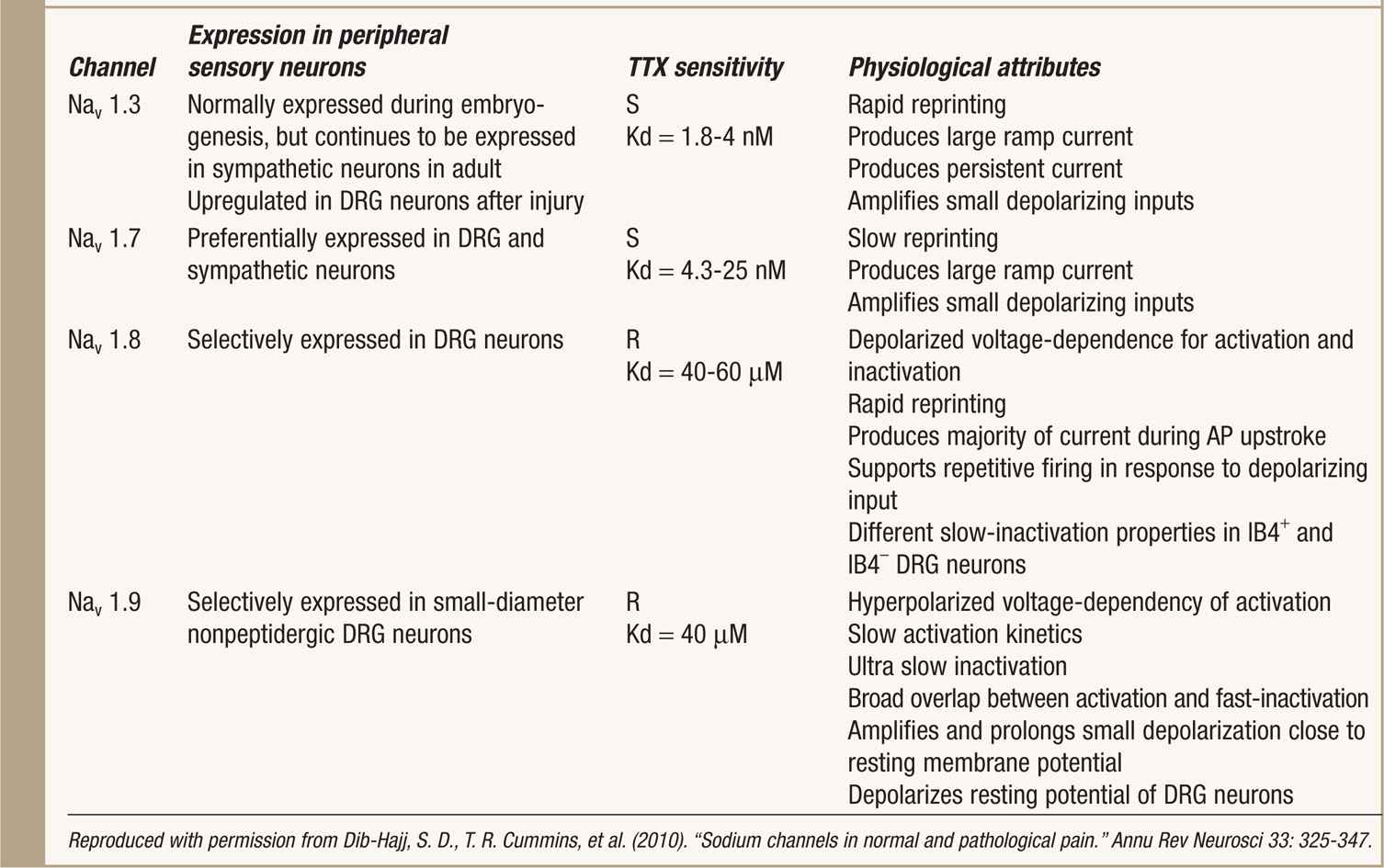
• Neuron specific NaV channels, such as NaV1.3, NaV1.6, NaV1.7, NaV1.8, and NaV1.9, are found on unmyelinated axons, nodes of Ranvier, and small dorsal root ganglion nociceptors (DGRs), and are pivotal for signal transmission along the pain axis.9
• Nav1.3, Nav1.7, Nav1.8, and Nav1.9 are preferentially expressed in peripheral sensory neurons and Nav1.3 is upregulated along pain-signaling pathways after nervous system injuries9 (Table 7-2).
• Nonneuronal NaV channels include NaV1.4, found in skeletal muscle, and NaV1.5, found in cardiac muscle. Further variation amidst individual NaV channels is seen by alternative splicing of genes.
TABLE 7-2. Sodium Channels Preferentially Expressed in Peripheral Neurons Add Reference and Acquire Permission
![]() An example of this is the NaV1.7 channel, in which 4 alternative splice variants are expressed in human DRG neurons.
An example of this is the NaV1.7 channel, in which 4 alternative splice variants are expressed in human DRG neurons.
![]() Furthermore, it has been found that this alternative splicing of the human NaV1.7 can specifically modulate the biophysical properties and cAMP-mediated regulation of this channel possibly influencing neuronal excitability and pain sensation.11,29
Furthermore, it has been found that this alternative splicing of the human NaV1.7 can specifically modulate the biophysical properties and cAMP-mediated regulation of this channel possibly influencing neuronal excitability and pain sensation.11,29
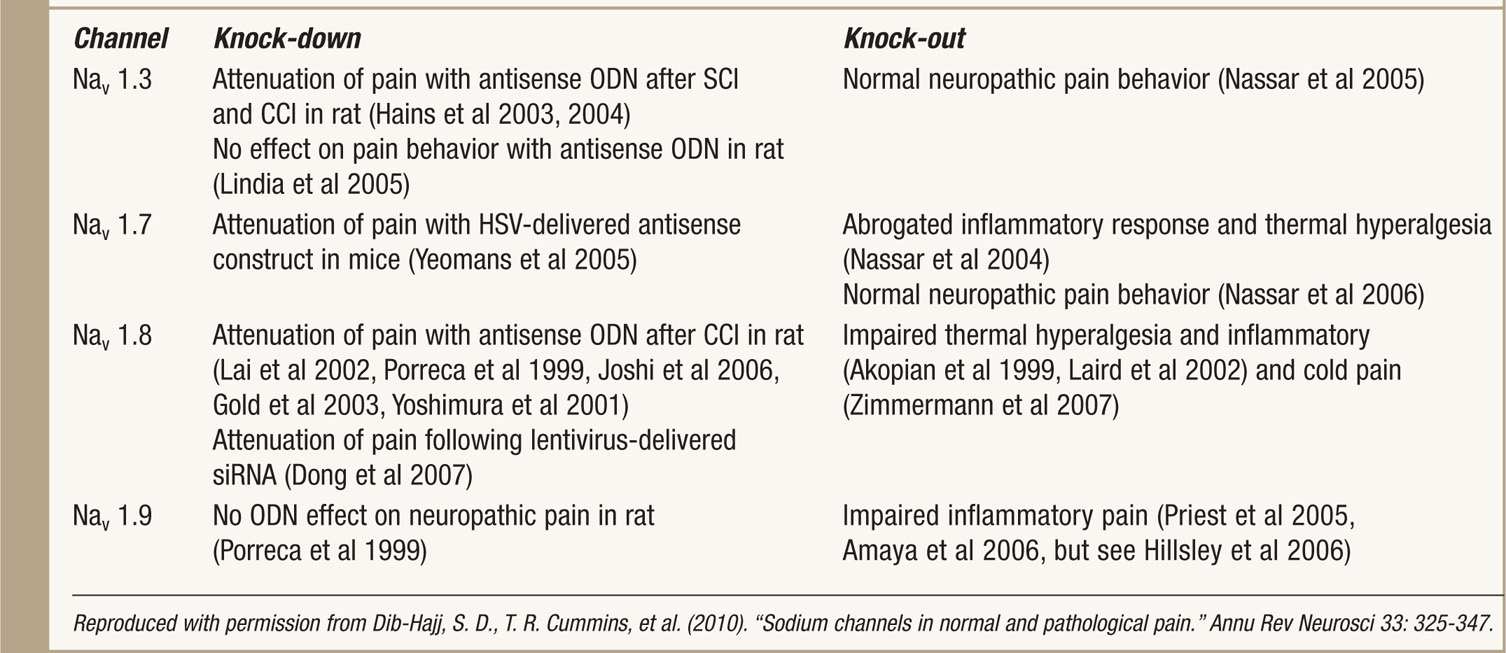
Individual isoforms are known to be particularly important in the pathophysiology of chronic pain syndromes9 (Table 7-3).
TABLE 7-3. Results of Knock-Down and Knock-Out Studies on Sodium Channels
FREQUENCY-DEPENDENT AND STATE DEPENDENT BLOCK
Because LAs bind preferentially to the NaV receptor in the activated-open state, an action potential must first be propagated to allow the receptor to assume this configuration.
The larger the activation potential and the more rapid the degree of firing, the greater the assumption of the activated-open state, and, hence the more rapid the block.
• This phenomenon is termed frequency-dependent block.
• It is also sometimes called “use-dependent,” when reference is made concerning the degree and rapidity of firing, and “state-dependent” block when reference is made concerning the activated-open state of the NaV channel having the greatest affinity for LAs.
• Of note, LAs do not just passively bind to one of the preexisting channel conformations, but may actively deform the channel, as described by the induced fit model.30,31
OTHER SITES OF ACTION
Since LAs have low potency, and are relatively nonselective for the NaV channel, they also bind and inhibit a variety of other channels, with nearly the same low affinity as for Na channels, and arguments have been put forward to include each of these receptors in the possible mechanism of action of LAs.10,32–38
To begin to appreciate whether or not these other channels play a role in the mechanism of action of local anesthetics in producing nerve conduction blockade, lidocaine 1 mM will reliably inhibit most Na channels, but will also inhibit (to a greater or lesser extent) nearly every other enzyme or ion channel.39
These additional binding sites could possibly contribute to LAs mechanism of action.32,39
Perhaps the main area of importance of receptors other than the NaV channels lies in their potential identification of binding sites and mechanisms that could contribute to local anesthetic production of neuraxial anesthesia, side effects, and clinical differences among the local anesthetics that cannot be explained by their relative affinities for Na channels.
MINIMUM CONCENTRATION
During the application of LAs for use in regional anesthesia, only a small fraction of these drugs will be bound to NaV channels.40
• In order for the intended anesthesia to ensure, a “critical length” of the nerve must be blocked.
• This length, though it varies based on nerve fiber type, generally exceeds 2 cm.
• For conceptual purposes, two or three consecutive nodes of Ranvier are approximately 1 cm.
• Note is made here that this length of greater than 2 cm for blockade is considerably larger than the commonly described 3 nodes of Ranvier. This length description is accurate except at very high LA concentrations.41
Cm is the term used to describe the minimum concentration of local anesthetic needed for conduction blockade. Several factors are known to influence Cm.
• A larger nerve fiber size requires a higher concentration of LA, whereas higher tissue pH and frequency of nerve stimulation decreases Cm.
• Each LA has its own individual Cm, however, as a generality motor fibers require twice the Cm as sensory fibers to be blocked.
• This observation contributes to the ability of LAs to provide sensory anesthesia without muscle paralysis.
Though animal studies have shown that lower volumes of more concentrated lidocaine produce shorter latencies and longer duration, it is the mass of drug delivered that determines the success of block.42 Accordingly, to achieve this mass effect, improvement in anesthesia is seen by both increasing volume or concentration.43,44
In comparing the relativity of ease of nerve impulse blockade, the most easily blocked nerve fiber types are preganglionic B fibers, which contributes to the sympathectomy seen with epidural anesthesia.
This concept describes differential conduction blockade, in which differing concentrations of LAs will block differing fibers.
• Following progressively higher concentrations, C fibers and small to medium A fibers are blocked; higher concentrations of LAs are generally required to block C fibers as compared to Aδ or Aβ fibers.45,46
• Blockade of these fibers at this concentration will block pain and temperature, however, touch, proprioception, and motor function will still be intact.
• For clinical applications, it is observed that the fastest onset and shortest duration of anesthesia occur with intrathecal or subcutaneous injections. This is compared to a slower onset and longer duration obtained with plexus blocks.
Changes in sensitivity to LAs can be seen in pregnancy in which more rapid onset of conduction blockade is seen with reduced doses.47 This is felt to be caused by protein binding characteristic changes in alpha-1-acid glycoprotein, the main binding protein of amide LAs, in which both total protein binding and protein concentration are reduced, caused by a nonprogesterone effect.48–50 It is also important to note that the spread of neuraxial anesthesia also increases during pregnancy, possibly related to an engorgement of epidural veins and the subsequent decreasing of the epidural fat space.10,51,52
PHARMACOKINETICS
LAs are weak bases with pKa ranging from 7.6 to 8.9 (see Table 7-1). This said, at physiologic pH (range from 7.35 to 7.45), less than half of LAs will exist in a nonionized, and, hence, a non-nerve penetrable form.
• As an example, procaine, with the highest pKa of LAs at 8.9, less than 5% of the molecule will exist in the nonionized form at physiologic pH.
• Conversely, LAs with pKas closest to physiologic pH, such as mepivacaine, pKa of 7.6, have the most rapid onset of action, having a more favorable ratio of nonionized molecules. It is the nonionized form of an LA that gains entrance into the cell though diffusion through the cell membrane.
• The onset of anesthesia generally slows with increasing LA lipid solubility.
ABSORPTION AND DISTRIBUTION
Several factors will influence the ultimate absorption into the systemic circulation of LAs.
These include:
• The site of injection, as LAs will have differing absorption rates at differing sites (Figure 7-7)
• Dosage
• Use of epinephrine
• Characteristics of the drug
• Mode of administration
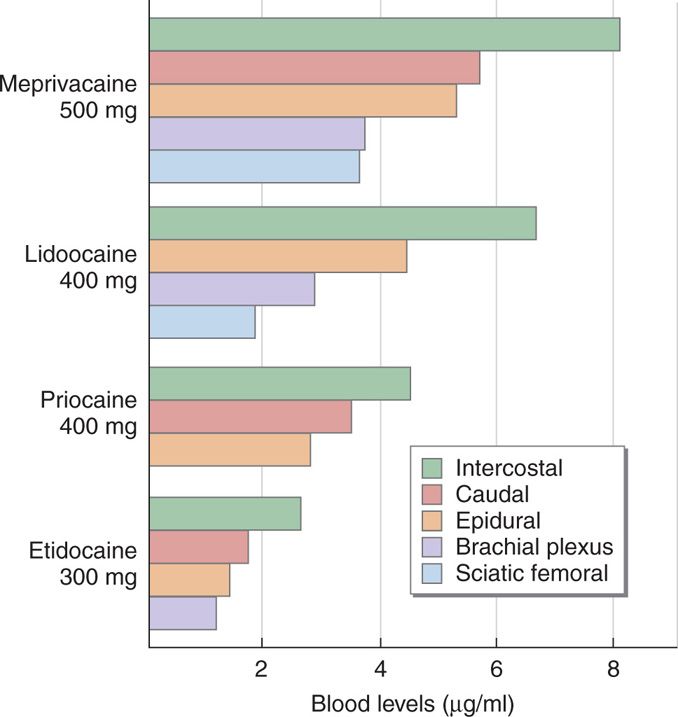
Figure 7-7. Peak plasma concentrations of local anesthetics are influenced by the site of injection for accomplishment of regional anesthesia.
Plasma concentration will be determined by
• Rate of tissue distribution and/or redistribution.
• Rate of clearance of the drug.53–55
Cardiac output is also a factor in tissue distribution and intercompartment clearance.56 Lipid solubility is important in redistribution, and is a primary determinant of intrinsic LA potency.
• LAs are ultimately cleared from the plasma via metabolism and excretion.
• Age and hepatic function will influence the absorption and plasma concentrations of LAs.
• Protein binding of LAs influence their distribution and excretion.
• Protein binding for LAs is in direct proportion to their lipid solubility, and inversely proportional to the plasma clearance of the drug.57–59
• Because of their slower metabolism (see below), amide LAs are more widely distributed in tissues compared with ester LAs.
It is well established that systemic absorption of local anesthetics correlates positively with the vascularity of the injection site, with ordering as follows:
intravenous > tracheal > intracostal > paracervical > epidural > brachial plexus > sciatic > subcutaneous6
In addition to site, duration of LA administration is also a factor.
PLACENTAL TRANSFER
Several factors influence the amount and degree of LA transfer between the maternal umbilical artery and the umbilical vein of the fetus:
• Protein binding of the LA
• Metabolism of the LA
• pH status of the fetus
In terms of protein binding, bupivacaine is approximately 95% protein bound, lidocaine is approximately 70% protein bound, and prilocaine is approximately 55% protein bound, and this order is in congruence with the lipid solubility of these compounds. When local anesthetics bind to proteins, they are unable to cross membranes via diffusion, as their free soluble fraction is not available. In reference to a drug’s ability to cross the placenta, only the free fraction in plasma is able to do so, and the amount that crosses the plasma through the placenta and into the fetal circulation is expressed as a maternal umbilical artery to fetal umbilical vein ratio (MUA/FUV).
As it follows, bupivacaine has an MUA/FUV of only 0.32, lidocaine 0.73, and prilocaine 0.85.60 Since ester LAs are rapidly hydrolyzed within seconds after they enter the systemic circulation, they are not available to transfer across the placenta in significant amounts. Additionally, once ester local anesthetics cross the placenta, they are rapidly degraded in the fetal circulation as well, further limiting their fetal toxicity.
Only the nonionized form of LAs can cross cell membranes. However, since LAs are weak bases, any acidic medium they encounter can cause ionization of the LA and prevent it from being able to diffuse passively across cell membranes. Such a scenario is possible in the fetus. When a fetus is stressed, secondary to a myriad causes normally related to hypoperfusion, the fetal pH can become acidotic. As such, any LAs that had diffused across the placenta in a nonionized form can become ionized in the fetus and, subsequently, trapped in the fetal circulation. This phenomenon has been described and termed the “ion trapping” of LAs. Clinically, given the widespread use of lidocaine in obstetric anesthesia, and lidocaine’s pharmacokinetic profile as described above, the risk of ion trapping is likely greatest with this LA.
CLEARANCE
Amide LAs are primarily metabolized in the liver. Their renal excretion in an unchanged form is minimal.
Ester LAs are rapidly metabolized via intravascular hydrolysis by pseudocholinesterase, and, if able to reach the liver, hydrolysis within the liver as well. Because this degradation is so rapid, pharmacokinetic studies are limited.
TOXICITY
True IgE mediated type I, or immediate, hypersensitivity allergies to LAs are associated with esters, not amides.
In comparison to the type I hypersensitivity reactions encountered with ester LAs, amide LAs can cause a type IV, or cell mediated hypersensitivity reaction. Accordingly, the intradermal injection patch test is the screening method used to determine if a specific substance causes allergic inflammation of the skin indicative of type IV hypersensitivity.
Because effects are more closely tied to the specific properties of local anesthetics, as a generalization, initial symptoms of local anesthetic toxicity
• Are lightheadedness and dizziness
• Followed shortly thereafter by perioral numbness
• Tinnitus
• Metallic taste in the mouth
• Progressive excitatory CNS effects can ensue and
![]() Include visual and auditory abnormalities
Include visual and auditory abnormalities
![]() Shivering
Shivering
![]() Muscular twitching
Muscular twitching
![]() In its highest level of CNS excitation, generalize tonic-clonic seizures
In its highest level of CNS excitation, generalize tonic-clonic seizures
• Furthermore, CNS depression can lead to respiratory depression or arrest.
In addition to allergic reactions, myotoxicity and neurotoxicity can be caused by all LAs if high enough concentrations are used. Transient neurologic symptoms (TNS) and cauda equina syndrome have been reported with the use of 2% and 5% lidocaine administered intrathecally.
In a study to determine the relative risk for developing TNS after spinal anesthesia with LAs, lidocaine was found to produce a greater incidence of TNS as compared to bupivacaine, prilocaine, procaine, and mepivacaine.61 Furthermore, in reference to TNS incidence, the lithotomy position appears to be a predisposing factor.
In an analysis of the myotoxic effects of LAs on muscle tissue, 1% procaine, 0.2% tetracaine, 0.5% lidocaine, and 0.75% bupivacaine, produced effects that were confined principally to the muscle fiber, whereas 2% chloroprocaine, 0.25% dibucaine, 0.5% lidocaine with 1:200,000 epinephrine and 2% piperocaine produced toxic effects on other tissue components of the muscles in addition to affecting muscle fibers.62
These effects were restored by regeneration and 30-day muscles showed relatively few long-term effects of the damage. However, it was concluded that procaine produces the least and bupivacaine the most severe muscle injury.62
In another study with ropivacaine, bupivacaine causes tissue damage of a significantly larger extent, and, additionally, induces apoptosis in skeletal muscle cells.63
When the iron within the hemoglobin molecule becomes oxidized, it changes from the ferrous (Fe2++) to the ferric state (Fe3+++), and methemoglobin results. Several LAs can produce methemoglobinemia, with prilocaine, usually administered via ELMA cream, and benzocaine, being the most recognized.
It is because of benzocaine’s association with methemoglobinemia, usually in the form of hurricane spray, that many hospital pharmacy systems in the US have stopped carrying this LA. However, it is important to note that both lidocaine and bupivacaine are also capable of forming methemoglobin as well.
Studies have revealed that the doses of bupivacaine required to produce convulsive activity and cardiac collapse are lower than that required of other LAs. In human studies, bupivacaine also causes CNS and cardiovascular toxicity at doses lower than that of either ropivacaine or levobupivacaine, which cause toxicity at approximately equipotent doses.64–67
Seizures induced by local anesthetics are usually self-limiting, and require maintenance of respiratory gas exchange and control of muscle contractions (eg, intubation, oxygenation, short-acting muscle paralysis).6
Drugs such as propofol, thiopental, midazolam, and diazepam are effective against these seizures.6,68–80 This also means that patients premedicated with midazolam or diazepam could manifest LA toxicity as cardiac collapse without seizures, especially with the use of bupivacaine.70
To state the apparent, should signs or symptoms of toxicity develop, one should cease the administration of LA.
As with all procedures, vital signs should be monitored in an effort to detect signs or symptoms of LAs as early as possible. Since it is not so much the total dosing of LA, but the rate of rise of LAs that most closely reflects risk for toxicity, care should be taken to avoid intravascular injections, especially intra-arterial injections that may expose the CNS to LA without a first pass metabolism through the liver or lungs. Additionally, injecting total doses of LA over multiple injections will also reduce the total plasma rate of rise of LA, and should be considered when feasible for interventional pain management procedures. Although the use of ultrasound, and other methods of direct visualization, for the placement of regional anesthesia and/or pain procedure blocks have allowed practitioners the ability to use lower volumes of LA, accidental intravenous or intra-arterial injection remains a risk.
In addition to the Advanced Cardiac Life Support recommendations for treatment of cardiac related symptoms secondary to the use of LA, there is evidence for the use of lipid emulsion in the treatment of bupivacaine cardiac toxicity.81 Until recently, it was unclear whether or not this treatment cross applies to other LAs is not known.82–85 However, a recent case report of a patient successfully resuscitated with lipid emulsion therapy after prolonged and intractable lidocaine toxicity perhaps demonstrates the need to consider lipid emulsion therapy in the advanced cardiac life support algorithm for lidocaine toxicity, as well as other lipid-soluble drug intoxications.86
METABOLISM AND UNIQUE PROPERTIES OF AMIDE LOCAL ANESTHETICS
Amide LAs are metabolized primarily in the liver via cytochrome P450 microsomal enzymes. Metabolites retaining LA activity and toxicity potential usually do so with lower potency than the parent compound, depending on the LA involved. Accordingly, of the amide LAs, prilocaine is metabolized the fastest, followed by lidocaine and mepivacaine, and, then followed by etidocaine, bupivacaine, and ropivacaine.
Since the metabolism of amide LAs are slower, compared with that of ester LAs, accumulation, and sustained plasma concentrations, are possible. As compared to ester LAs, this leads to a higher risk of systemic toxicity with the amide LAs.
Lidocaine. Lidocaine undergoes oxidative dealkylation in the liver to monoethylglycinexylidide, which is then hydrolyzed to xylidide.
Monoethylglycinexylidide is an active metabolite with a long elimination half-time. It has about 80% of the activity of lidocaine in protecting against cardiac dysrhythmias.
Etidocaine. Less than 1% of etidocaine is secreted in the urine unchanged. Etidocaine causes a motor block before blocking sensory nerves. Additionally, the motor block can out last the sensory block, giving rise to a very unpleasant experience of a patient being in pain, however, lacking motor ability.87,88 Because of this, it is rarely used in current clinical practice.
Prilocaine. Prilocaine can be metabolized to an oxidizing metabolite, orthotoluidine. Orthotoluidine can oxidize the iron molecule in hemoglobin to form methemoglobin. When methemoglobin levels reach 3 to 5 mg/dL, oxygen carrying capacity can be reduced such as a patient may appear cyanotic. Typically, prilocaine doses >600 mg can cause this. Methemoglobinemia can be reversed with the use of methylene blue.
Mepivacaine. Mepivacaine also lacks vasodilator activity, and, as such, is an alternative when desiring to use an LA without epinephrine in a given clinical scenario. If used in neonates, the clearance of mepivacaine can be prolonged.
Given mepivacaine’s fast onset, mepivacaine has been mixed with bupivacaine in an attempt to decrease the speed of onset of the block. However, data is lacking as to the onset, duration, or quality of the ensuing block. This said, toxicity seems added, as the seizure dosages and arterial plasma levels for drug mixtures have been investigated and found as such.89
Bupivacaine. Bupivacaine is metabolized effectively in humans, as less than 1% of bupivacaine is excreted unchanged in the urine.90 Four metabolic processes have been proposed: (1) aromatic hydroxylation, (2) N-dealkylation, (3) amide hydrolysis, and (4) conjugation.91,92 Ring 4′-hydroxylation and N-dealkylation, and subsequent conjugation, are minor metabolic pathways in humans, whereas preliminary results indicate that bupivacaine may be resistant to amide hydrolysis.90,92,93
Its toxicity includes seizures and refractory cardiac arrhythmias.94 The plasma threshold in which these toxic effects occur varies between 2 and 5 ug/L. The reason why such a wide range is given, is because the toxic effects of bupivacaine rely more on the rate of rise of the serum concentration, then on the absolute amount of the drug administered.
Levobupivacaine. Levobupivacaine is the S-enantiomer of bupivacaine. In relation to bupivacaine, its duration of action is longer, it possesses less vasodilation compared with bupivacaine, and it is approximately 13% less potent than racemic bupivacaine.
Ropivacaine. Ropivacaine is largely excreted in the urine (86%), however, unchanged ropivacaine is excreted by the kidney only approximately 1%. It is mainly metabolized by the liver cytochrome p450 system into 2, 6-pipecoloxylidide and 3-hydroxyroropivacaine, both significantly less potent than the parent compound.
METABOLISM AND UNIQUE PROPERTIES OF ESTER LOCAL ANESTHETICS
As a whole, ester type local anesthetics are hydrolyzed rapidly in the blood by cholinesterase enzymes, particularly pseudocholinesterase, and by a smaller amount of these enzymes in the liver. In general, the rate of hydrolysis varies among the ester type local anesthetics, with the order, from the quickest to the longest, being Chloroprocaine, procaine, and tetracaine. The longer the drug is available to exert its effect on the body, the longer it is able to produce toxic effects as well. Accordingly, toxicity is inversely related to the rate of hydrolysis. Procaine and benzocaine are metabolized to p-aminobenzoic acid (PABA).
2-Choloroprocaine
Choloroprocaine is perhaps the only ester type local anesthetic still used commonly in current clinical settings. Choloroprocaine undergoes oxidative decomposition, and, as such, numerous formulations have been tried to decrease this event. Currently, it is manufactured in colored vials to reduce the entry of light that can catalyze this event.
In the pregnant state, in vitro plasma half-life is 21 seconds in maternal blood and 43 seconds in fetal blood, hence making fetal effects virtually nonsignificant. Even in cases of atypical cholinesterase were individuals are homozygous for the atypical pseudocholinesterase, the half-life is only prolonged to about 2 minutes.
Aside from ester, local anesthetics not being used in IV regional blocks because they are broken down rapidly in the blood, there has also been noted the incidence of thrombophlebitis with the use of choloroprocaine.
Procaine
Less than 50% of procaine is excreted in the urine, whereas the rest is metabolized to both paraaminobenzoic acid and diethylaminoethanol. Paraaminobenzoic acid is excreted unchanged in the urine, whereas diethylaminoethanol is further metabolized. Allergy to procaine is usually due to paraaminobenzoic acid, and not procaine itself. PABA is not associated with symptoms of systemic toxicity.
Procaine, having vasodilator properties, it is often used in conjunction with epinephrine.
Benzocaine
Benzocaine is the ethyl ester of PABA, and, as such, PABA is formed as a metabolite in its metabolism. Considering its nonionized form, it is ideally suited for topical uses as it can more easily penetrate epithelial surfaces with a rapid onset and duration of 30 to 60 minutes. As such, a common use is for awake tracheal intubation, in which single sprays of formulations of 20% benzocaine delivers the recommended dose of 200 to 300 mg. Benzocaine has the potential to form methemoglobinemia should doses exceed the recommended dose. Another popular formulation is cetacaine, which is a combination of 14% benzocaine, 2% tetracaine, and 2% butamben.
Tetracaine
Like all esters, tetracaine is metabolized by hydrolysis by cholinesterases, primarily in the plasma and to a lesser extent by the liver. Its metabolites include a PABA containing metabolite diethylaminoethanol.
In current medical practice it is occasionally used in spinal anesthesia.
Cocaine
Cocaine is metabolized by plasma and liver cholinesterases into water-soluble metabolites that are excreted in the urine. About 1% of the drug is excreted unchanged in the urine. With the primary metabolic event being hydrolytic ester cleavage, the main metabolite is benzoylecgonine (BE), with lesser metabolites being ecgonine methyl ester (EME) and ecgonine, and additional minor metabolites including norcocaine, p-hydroxycocaine, m-hydrococaine, p-hydroxybenzoylecgonine (pOHBE), and m-hydroxybenzoylecgonine.95 These metabolites are used for assays to detect cocaine use in suspected patients. In general, contingent on hepatic and renal function, benzoylecgonine can be detected as soon as 4 hours within the urine, and could be detectable for up to 8 days.
Accordingly, any disease or physiological states, such as pseudocholinesterase deficiency or pregnancy can prolong the elimination life of cocaine. Cocaine in addition to being a local anesthetic, is a serotonin-norepinephrine-dopamine reuptake inhibitor, with affects on the mesolimbic reward pathway that renders it addictive.96
Given cocaine’s strong inherent ability to produce intense vasoconstriction, in modern medical practice, it is predominantly used for nasal and lacrimal duct surgery and as an adjunct in nasal fiberoptic intubation.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


