Inflammation presumably evolved as a process to achieve two principle benefits—wound healing and defense against microbiologic invasion. When insults are relatively small (i.e., elective groin hernia wound, facial acne), then the physiology and cellular functions of these two processes can be considered local, somewhat distinct, with little effect on the rest of the body (little systemic effect). But when insults are large (i.e., bilateral femur fractures with bilateral pulmonary contusions, perforated sigmoid diverticulitis), then the similarities between wound healing and host-defense mechanism become more apparent and associated with more evidence of systemic alterations.
First, here are descriptions of the more local physiology and cellular functions of wound healing and host defense, followed by descriptions of how wounds and microbiologic invasion result in systemic inflammation.
Wound Healing
When a wound is in the skin and subcutaneous tissue (the most common wound studied), the epidermal barrier is broken and keratinocytes release pre-stored interleukin-1 (IL-1). The subsequent response is bleeding and coagulation. Platelet activation results in the release of important chemoattractants [i.e., epidermal growth factor (EGF), platelet-derived growth factor (PDGF), transforming growth factor-beta (TGF-β)].
Damaged blood vessels initially constrict, but this is soon followed by vasodilation and increased capillary permeability secondary to the action of agents such as prostaglandin E2, prostacyclin, histamine, serotonin, and kinin. This vascular response results in the accumulation of protein-rich edema fluid (exudate). Leukocyte cells adhere to the damaged and leaky vessels.
Attracted by chemoattractants such as PDGF and IL-1, polymorphonuclear cells (PMNs) are the first leukocytes to migrate to the inflammatory site (within minutes if the circulation is good). PMNs serve to phagocytize dead tissue and foreign objects. Removal of bacteria may be assisted by opsonins and preformed antibodies. PMNs produce proteases and intracellular oxygen radicals that are critical for beneficial PMN activity. Besides proteases and oxygen radicals, PMNs can release IL-1. IL-8 is a potent PMN attractant produced by many cell types after incubation with IL-1 and tumor necrosis factor (TNF). The PMNs last only for hours.
Lymphocytes are the next cell to migrate into the wound. The role of the lymphocyte is less well understood, but depletion of T cells results in impaired wound healing.
Subsequently (within hours), tissue macrophages and circulating monocytes are attracted by substances such as PDGF, TGF-β, and IL-1, migrate into the injured area, and last for days to weeks. Wounds can heal without PMNs but not without macrophages that regulate most of the continuing stages of inflammation and wound healing through mediators such as IL-1, PDGF, TGF-β, TGF-α, and fibroblast growth factor (FGF).
Fibroblasts migration and angiogenesis begin next. Fibroblasts are influenced by IL-1, PDGF, TGF-β, and TGF-α; angiogenesis is influenced by TGF-β, TGF-α, and EGF. The combined process of fibroblast proliferation and capillary budding produces a wound that is granular in appearance (granulation tissue), very vascular, and quite friable. Certain fibroblasts—myofibroblasts—have smooth muscle contractile elements that contract and diminish the area of a wound. In general, wound contraction continues until the lining cells from each edge of a wound meet (epithelialization for the skin). Therefore, slight contraction will follow wounds that are closed primarily, that is, with the lining edges apposed. Much contraction may occur in secondary healing, especially when wound edges remain widely separated for days to weeks.
Fibroblasts make collagen, usually accelerating at five days after tissue damage. Before this, fibrin is the principle wound substance besides sutures that holds a wound together. Collagen synthesis is also influenced by IL-1, PDGF, TGF-β, and EGF. Since the macrophage is an important source of these factors, mechanisms that increase (glucan administration) and decrease (the combined effect of hypoxia and endotoxin) macrophage function may result in increased or decreased wound strength, respectively.
A summary of cellular activity in wound healing is provided in
Tables 4.1 and
4.2 (
1,
2).
Local Host Defense
Local host-defense mechanisms have been principally studied as the response to microbiologic challenge. Both an
innate immune capacity (defenses that lack immunologic memory) and an
acquired capacity (defenses with immunologic memory) are components of local host defense (
Tables 4.3 and
4.4).
The first mechanism of local innate host defense is surface barrier function, best understood as the effect of the intact skin. Penetration through a barrier results in further stimulation of the innate response.
The polymorphonuclear leukocyte (PMN, neutrophil) is the earliest cell component that responds to pathogen penetration. The steps related to PMN infiltration and action are listed in
Table 4.3. PMNs serve to phagocytose organisms and/or debris, kill microorganisms, and release enzymes and reactive oxygen species to enhance control of damaging materials. Complement coating of organisms enhances phagocytosis (
3,
4,
5,
6,
7).
PMN activation also results in the release of cytokines (IL-1, IL-6, TNF-α, IL-8, IL-12), toxic oxygen radicals, peroxides, nitric oxide (NO), and lipid mediators of inflammation such as prostaglandins and leukotrienes, as well as platelet activating factor (PAF) (
5).
Macrophage infiltration follows PMN activation and accomplishes many of the same microbe suppression activities. In addition, macrophages are responsive to pathogen-associated molecular structures (PAMP) that are shared by entire classes of pathogens, such as lipopoly-saccharides on Gram-negative bacteria. Macrophage recognition of a PAMP triggers activation before proliferation at the site (
8). The macrophage also serves as an antigen-presenting cell for stimulation of the acquired immune system. However, the dendritic cell, also responsive to PAMP, is a more potent antigen-presenting cell that migrates to local lymph nodes to engage T cells (
7).
The principal PAMP receptors are called toll-like receptors, first described in drosophila, and now considered a primary mechanism for activation of nuclear factor-kappa B (NF-κB), an important step in the production of cytokines and co-stimulatory molecules (
8,
9,
10).
Natural killer cells can recognize “non-self” cells by absence of the major histocompatibility complex (MHC) class I. Since MHC class I is present on nucleated cells, most are not subject to threat. However, infection can alter the expression of MHC class I on infected cells and result in killer cell destruction (
7,
11).
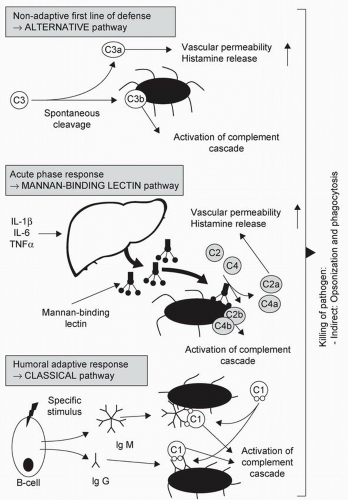
Complement is another component of the innate immune system that can be activated via three pathways (
Figure 4.1). The classical pathway is activated by antibodies [immunoglobulin (IgG, IgM] binding to antigen and is part of acquired immunity. The mannan-binding lectin (MBL) pathway is activated by binding of MBL, an acute-phase reactant secreted by the liver, to bacteria or virus surface components. In the alternative pathway, the complement component C3 becomes “spontaneously activated” to C3b that then binds to the surface of a pathogen and promotes opsonization. Overall, activation of complement results in opsonization, recruitment of inflammatory cells, and direct killing of pathogens (
5).
Small amounts of tissue injury and infection can presumably result in principally autocrine and/or paracrine effects that do not result in any cellular activity distant from the site of the stimulus. However, sufficient tissue injury and infection initiates an endocrine effect such
that mediators of inflammation cause systemic alterations that can be adaptive or maladaptive, usually depending on the magnitude of insult and, sometimes, the magnitude of the individual’s response (see section on “Severe Systemic Inflammation”).
Adaptive systemic effects that appear to enhance host defenses are fever (secondary to IL-1, IL-6, and TNF-α) and liver synthesis of acute phase reactants [i.e., C-reactive protein (CRP) and MBL], which are enhanced by these same cytokines (
5).
As with wound healing, the local response to pathogen penetration can result in complete resolution of tissue injury with little scar formation. However, incomplete resolution can result in ongoing inflammation, granulation tissue formation, and fibrosis, collectively called an abscess. An abscess may or may not be associated with systemic alterations, again dependent on the magnitude of the threat as well as that of individual response.
Acquired immunity depends on the presentation of foreign antigens to T cells. Dendritic cells, macrophages, and B cells serve to present antigen to unstimulated T cells. Antigens that gain access to the circulation can be captured by antigen-presenting cells in the spleen and stimulate T cell activation at that site. The naïve T cells can then differentiate into Th1 cells that augment cell-mediated immunity or Th2 cells that augment humoral immunity. This differentiation is linked to a complex interaction of inflammatory mediators. Full activation of acquired immunity takes several days (
5).
In addition to these inflammatory cell activities, a local site of infection is characterized by arteriolar vasodilation, increased blood flow, increased capillary permeability, exudation of plasma, and pain. This results in the complaint of pain (dolor), and the findings of erythema (rubor), edema (tumor), and warmth (calor).