Chapter 76 Inborn Errors of Metabolism
Metabolism can be defined as the sum of all biochemical processes that convert food to smaller molecules and energy for the purposes of structure and function. An inborn error of metabolism (IEM) is an inherited deficiency of any critical step in metabolism. Although genetic deficiency of catalytic enzymes in intermediary metabolic pathways is the classic paradigm for IEM, the pathophysiology of metabolic disorders may involve abnormalities of any number of cellular processes, including transmembrane transport, cell signaling, cell differentiation and development, energy production, and others. Many IEMs are individually rare, although a few, including phenylketonuria (PKU) and medium-chain acyl-coenzyme A (acyl-CoA) dehydrogenase deficiency (MCADD), a defect in fatty acid oxidation, exhibit a population incidence approaching 1:10,000 live births.1,2 Specific IEMs may be more common in certain ethnic groups with a history of relative reproductive isolation. Collectively, the population incidence of all IEMs may approach 1:1500 live births, depending upon how broadly IEM is defined. Many IEMs are associated with catastrophic illness necessitating advanced life support. Although IEMs may present very rarely within the professional lifetime of the average medical practitioner, critically ill children with IEMs will not be uncommon visitors to the pediatric intensive care unit (ICU), especially in a tertiary care center.
Other published textbooks on the diagnosis and treatment of IEM provide an exhaustive list of known disorders.3,4 Rather than recapitulate an encyclopedia of possible diseases, this chapter presents a diagnostic rationale based upon specific clinical symptom complexes that are likely to occur in the critically ill child. Algorithms for the differential diagnosis of specific clinical scenarios are given in support of this rationale. Symptoms often begin during early infancy in the biochemically most-severe IEMs; naturally, these IEMs with neonatal onset are the focus of our discussion in this chapter. However, “milder” or late-onset variants of virtually every IEM have been described, with onset of symptoms occurring at all ages, even during adulthood. Some IEMs uniformly present after the neonatal period; age of symptom onset (late infancy, childhood, or adulthood) often is an important clue to the specific diagnosis. The clinical presentation, diagnostic workup, and treatment of neonatal onset disorders provide a paradigm for the evaluation and management of possible IEM in a child of any age.
Pathophysiology of Inborn Errors of Metabolism
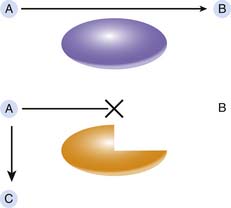
Under the classic paradigm, an IEM is associated with deficiency of a specific protein, often a catalytic enzyme, involved in a critical metabolic pathway (Figure 76-1). This deficiency leads to a block in the pathway and the accumulation of the enzyme substrate. In this model, three distinct pathogenic mechanisms are possible proximate causes of the symptoms associated with an IEM. The specific pathogenic mechanism involved in any given IEM dictates the appropriate treatment strategy. First, accumulation of the substrate may lead to toxic effects at very high levels; successful therapy requires effective elimination of the substrate or a method to block its toxic effects. An appropriate example for this mechanism is PKU, in which elevated phenylalanine levels adversely affect neuronal development, and the reduction of tissue phenylalanine content through dietary phenylalanine restriction largely prevents the major clinical features of PKU.5 Second, deficiency of the reaction product, should it be a critically important metabolite, may lead to disease. Supplementation with the essential metabolite, if possible, may cure the disease. Biotin is a required cofactor for four distinct carboxylase enzymes. Deficiency of free biotin develops in the face of genetic biotinidase deficiency and leads to symptoms of multiple carboxylase deficiency. Supplementation with oral biotin completely prevents the clinical manifestations of biotinidase deficiency.6 The final pathogenic mechanism involves the conversion of the enzyme substrate, through normally quiescent alternative pathways, to toxic secondary metabolites. Elimination or decreased production of these secondary metabolites may improve disease symptoms. For example, tyrosinemia type I (fumarylacetoacetate hydrolase [FAH] deficiency) is associated with recurrent attacks of abdominal pain and paresthesias reminiscent of acute intermittent porphyria. The accumulating substrate, fumarylacetoacetic acid, is converted through secondary pathways to succinylacetone, and succinylacetone in turn inhibits the heme synthetic pathway and causes porphyria-like symptoms. Pharmacologic inhibition of the tyrosine catabolic pathway proximal to the block at FAH decreases the production of fumarylacetoacetic acid and succinylacetone and alleviates the pathology associated with these toxic compounds.7
Signs and Symptoms of Inborn Errors of Metabolism
Clinical signs and symptoms frequently associated with IEMs are listed in Box 76-1. The symptom repertoire of the critically ill infant is limited, and the clinical presentation of metabolic disorders often is nonspecific. It is for this reason that the diagnosis of an IEM may be easily missed. To maintain maximum diagnostic sensitivity for IEMs, the clinician must maintain a high level of suspicion and be willing to initiate screening metabolic laboratory studies with little provocation. As was true for appendectomies in the era prior to the advent of ultrasound-based diagnosis of appendicitis, a certain number of nondiagnostic metabolic laboratory workups in sick children must be performed to ensure ascertainment of individuals with inherited metabolic disorders. In particular, IEM should be a strong diagnostic consideration in any neonate who has become catastrophically ill following a period of normalcy. This presentation may be clinically indistinguishable from bacterial or viral sepsis, and the nonspecific supportive therapy provided to potentially septic infants (fluid and glucose administration) may alleviate the symptoms and mask the presence of an IEM. Diagnostic metabolic laboratory studies are most likely to provide definitive information if performed on clinical samples obtained at initial presentation and before any therapy is initiated. Failure to obtain the necessary specimens at this time may miss an important diagnostic window of opportunity. Many children with IEM have been saved initially by intensive but nonspecific treatment but then suffered clinical relapse or even death in the absence of the correct diagnosis. Certainly, the possibility of an IEM should be considered in any child for whom the clinical picture suggests sepsis but the laboratory evaluation for sepsis is negative. Unfortunately, bacterial sepsis is often a complicating factor in critically ill children with IEM. For example, Escherichia coli infection (including pyelonephritis, bacteremia, or meningitis) is frequently detected at presentation in infants with galactosemia. The astute clinician remains ever vigilant for the signs and symptoms that may suggest an inherited metabolic disorder.
BOX 76–1 Signs And Symptoms of Inborn Errors of Metabolism
Acute illness after period of normal behavior and feeding (hours to weeks)
Recurrent decompensation with fasting, intercurrent illness, or specific food ingestion
Persistent or recurrent vomiting
Hepatomegaly or liver dysfunction
Unexplained hemorrhage or strokes
Developmental delay with unknown etiology
Seizures, especially if seizures are intractable
Chronic movement disorder (ataxia, dystonia, choreoathetosis)
Family history of unexplained death or recurrent illness in siblings
Recurrent episodes of vomiting and dehydration in response to fasting or intercurrent illness are an important clue to IEM in older infants and children. Feeding difficulties and failure to thrive are common chronic complications. Children with unexplained hypotonia, developmental delay, or movement disorder should be evaluated for possible IEM. Inherited neurodegenerative disorders, such as the lysosomal storage diseases, stereotypically cause developmental regression, specifically loss of previously attained developmental milestones. Several IEMs are associated with major physical anomalies (Table 76-1). When present, these anomalies are exceedingly valuable in suggesting a specific diagnosis and directing the diagnostic evaluation. More commonly, the child with IEM is morphologically normal, and the presenting symptoms are nonspecific. The clinician must then rely upon screening laboratory tests to evaluate the potential for IEM.
Table 76–1 Physical Anomalies Associated with Inborn Errors of Metabolism
| Dysmorphic facial features | |
| Structural brain anomalies | |
| Macrocephaly | |
| Cataracts | |
| Lens dislocation | |
| Pigmentary retinopathy | |
| Renal cysts | |
| Ambiguous genitalia | |
| Skeletal abnormalities | |
| Hair or skin abnormalities |
Laboratory Evaluation of Suspected Inborn Errors of Metabolism
Abnormal results of routine laboratory studies may provide clues to the presence and type of IEM (Table 76-2). Highly informative but sometimes subtle laboratory abnormalities are often overlooked, especially in a busy ICU or hospital ward. For instance, a clinically relevant newborn screening result may have been sent to the primary care provider or birth hospital but not efficiently communicated to the ICU, in a different hospital, to which the now critically ill infant has been admitted. It is imperative to verify the infant’s screening results with the primary care provider or newborn screening laboratory (Box 76-2). Calculation of the anion gap, another example of a routine and highly informative result, is key to the differential diagnosis of metabolic acidosis. The absence of urine ketones in hypoglycemic children older than 2 weeks strongly suggests impaired ketogenesis as a consequence of either hyperinsulinism or fatty acid oxidation disorder. On the other hand, fatty acid oxidation and ketogenesis are incompletely developed in neonates. The presence of ketones in the urine of infants younger than 2 weeks is very unusual even during fasting or hypoglycemia and suggests the presence of an unusual keto acid, such as those excreted in maple syrup disease or the organic acidemias. Keto acids, organic acids, and sugars such as galactose or fructose increase urine specific gravity. Urine specific gravity greater than 1.020 in any neonate or in a well-hydrated older child suggests the unexpected presence of an osmotically active substance. Routine urinalysis at many hospitals may not include use of the Clinitest to detect reducing substances. Urine Chemstrips utilize a colorimetric glucose oxidase-based method to specifically detect glucose. This test does not react with any other sugar (galactose or fructose). However, some bedside glucose monitoring systems do react with galactose or fructose; inappropriately elevated capillary blood “glucose” accompanied by a normal venous glucose as measured by chemistry analyzer suggests the presence of a sugar other than glucose in the blood. A comatose infant with a blood urea nitrogen (BUN) level below the limits of detection may have an inherited defect in the urea cycle. Blood ammonia measurement is crucial to confirming that suspicion. Failure to check the blood ammonia level has caused missed diagnoses, failure to appropriately treat hyperammonemia, and further morbidity and mortality in comatose infants with urea cycle disorders or organic acidemias. Finally, bacterial sepsis and meningitis are more common causes of severe lethargy and coma in infants than is IEM, but bacterial infection may also be a complicating feature in severely ill infants with IEM. Infants with galactosemia, for example, are particularly prone to pyelonephritis, bacteremia, sepsis, or meningitis, often with E. coli, as noted above. Antibiotic therapy without diagnosis and specific treatment of the underlying disorder may be useful in the short term but does not mitigate long-term IEM-specific effects.
Table 76–2 Initial Laboratory Evaluation of Suspected Inborn Errors of Metabolism
| Laboratory Test | Abnormality | Disorder |
|---|---|---|
| Complete blood count | ||
| Serum electrolytes | Metabolic acidosis | |
| Blood gas | ||
| BUN | Low or undetectable BUN (with hyperammonemia) | Urea cycle disorders |
| Transaminases (ALT, AST) | Liver dysfunction | |
| Total and direct bilirubin | Hyperbilirubinemia | |
| Serum uric acid | Hyperuricemia | |
| Blood ammonia | Hyperammonemia | |
| Blood lactate | Lactic acidemia | |
BUN, Blood urea nitrogen; FAO, fatty acid oxidation; MSUD, maple syrup urine disease.
Box 76–2 Screening Metabolic Laboratory Studies for Children with Suspected Inborn Errors of Metabolism
Suspicion of an IEM based upon clinical and routine laboratory findings should initiate specialized biochemical testing (Table 76–3). In the case of severely ill infants or when the clinical suspicion of IEM is very high, consultation with a biochemical geneticist, even if only by phone, is strongly advised to help direct the laboratory investigation and initial therapy. When the clinical presentation is nonspecific, that is, catastrophic illness in a previously well child without signs of any particular IEM, the “shotgun” diagnostic evaluation should minimally include plasma amino acid analysis, urine organic acid analysis by gas chromatography-mass spectrometry, and a so-called urine metabolic screen. The battery of qualitative assays included in a urine metabolic screen differs among laboratories, and the ordering clinician should be aware of which tests and disorders are included in the repertoire of the diagnostic laboratory chosen. Furthermore, although diagnostic laboratories in the United States must meet Clinical Laboratory Improvement Amendments requirements and often are accredited by the College of American Pathologists, the testing methodologies used, the quality of diagnostic testing for IEM, and more problematically, the availability of laboratory-associated consultants with experience in the diagnosis and treatment of IEM vary widely among laboratories. Although the ability of clinicians to direct clinical specimens toward specific diagnostic laboratories may be inhibited by contractual arrangements between the hospital and large referral laboratories, the critically ill patient is best served by diagnostic evaluation carried out in a timely manner by an experienced biochemical genetics laboratory, with laboratory staff available by phone for expert consultation on interpretation of test results.
Table 76–3 Biochemical Genetic Laboratory Studies
| Specimen | Test | Disorder |
|---|---|---|
| Blood | Plasma amino acid analysis | Aminoacidopathies |
| Plasma carnitine | ||
| Plasma acylcarnitine profile | ||
| Serum transferrin electrophoresis | Congenital disorders of glycosylation | |
| Urine | ||
| Organic acid analysis | ||
| Acylglycine profile | ||
| Quantitative mucopolysaccharide measurement and electrophoresis | Mucopolysaccharidoses | |
| Qualitative sulfites (Sulfitest) or quantitative sulfocysteine | ||
| Quantitative succinylacetone | Tyrosinemia type 1 | |
| Quantitative purines | Purine synthesis disorders |
PKU, Phenylketonuria; FAO, fatty acid oxygenation; MSUD, maple syrup urine disease.
The specific clinical presentation or specific screening laboratory findings may direct the intensivist or biochemical geneticist to order other more specialized metabolic tests (see Table 76-3). These analyses may provide diagnostic confirmation for specific disorders and supportive evidence alone for others. For several IEMs, confirmation of diagnosis may require enzyme activity analysis in tissue (red blood cells, lymphocytes, cultured skin fibroblasts, liver, or skeletal muscle depending upon the specific disorder in question) or molecular DNA testing for a specific gene defect. In general, these tertiary tests, which are often difficult, labor-intensive, and expensive, should be ordered following consultation with a biochemical geneticist. In some instances, confirmatory diagnostic biochemical or molecular tests are available only through specialized research laboratories.
Postmortem Evaluation of a Child with Suspected Inborn Errors of Metabolism
Some IEMs, particularly those exacerbated by fasting, may present as sudden infant death. For many IEMs, acute metabolic compensation may be rapid and lethal despite intensive medical intervention. The time after clinical presentation but prior to death may be insufficient to execute an adequate metabolic evaluation. Disease diagnosis is still possible postmortem and is important for fully understanding the cause of death and determining recurrence risk in the family. A protocol for postmortem evaluation of an infant or child with suspected IEM is given in Box 76-3. Many of the biochemical genetic analyses recommended for acutely ill children are still valid on postmortem specimens. Valuable information may be learned from amino acid, carnitine, and acylcarnitine analyses in blood and from metabolic screening and organic acid analysis in urine. However, collection of blood and urine may not be possible postmortem, especially if the autopsy is performed many hours after death. In these instances, metabolic testing may be obtained on alternative specimens such as vitreous humor or bile. In the event that screening biochemical studies suggest a specific diagnosis, disease confirmation by enzyme analysis in tissue is highly desirable. Many enzymes can be assayed in cultured fibroblasts; viable fibroblasts may be cultured from skin or Achilles tendon samples obtained as late as 24 hours after death. Biopsies of other organs may be necessary for analysis of certain other enzymes. Muscle, liver, and kidney specimens may be obtained postmortem for enzymatic analysis, but most enzymatic activities in solid organs deteriorate rapidly following death. Collection of specimens as soon as possible after death is critical for valid enzyme analyses.
Box 76–3 Postmortem Biochemical Genetic Evaluation
Modified from Steiner RD, Cederbaum SD: Laboratory evaluation of urea cycle disorders. J Pediatr 138 (Suppl 1):S21-29, 2001.
Analyses are most reliable if obtained within 6 hours after death.





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


