Chapter 62 Hypoxic-Ischemic Encephalopathy
Pathobiology and Therapy of the Post-Resuscitation Syndrome in Children
In the past decade, important clinical trials demonstrating neuroprotective effects of therapeutic hypothermia in adults with cardiac arrest and neonates at risk for hypoxic ischemic encephalopathy (HIE) make this an exciting era for pediatric intensivists.2–5 These trials (discussed later in this chapter) showing that induced hypothermia can prevent neurologic damage after cardiac arrest provide a window of opportunity for the application of pharmacologic or other interventions in what remains an important clinical problem with exceedingly high mortality and disappointingly high morbidity—cardiac arrest in children. At minimum, these trials have spurred renewed interest in determining whether therapeutic hypothermia is efficacious in infants and children after cardiopulmonary arrest, and were crucial in the implementation of a National Institutes of Health-funded multicenter clinical trial of therapeutic hypothermia after pediatric cardiac arrest in the United States. Although these “breakthroughs” and increasing depth and breadth of the understanding of mechanisms of HIE provide hope, challenges remain as by and large the likelihood of intact neurologic survival in children who suffer cardiopulmonary arrest remains unacceptably small.6,7 A proven effective treatment protocol that reliably prevents HIE and improves neurologic recovery after cardiopulmonary arrest in infants and children remains elusive. Despite numerous advances in critical care medicine, the optimal supportive care to maintain neuronal function and maximize recovery after cardiorespiratory arrest is in and of itself controversial.
This uncertainty arises from the pathobiologic complexity of cerebral injury and from the limitations in our clinical ability to monitor key metabolic and physiologic parameters in the brain. Clinical stumbling blocks in the history of “brain resuscitation” have also slowed progress in our understanding of HIE after cardiopulmonary arrest. Historically, this entity was largely ignored as a specific disease process. Brain resuscitation was dealt with as a single therapeutic paradigm regardless of the etiology.8 This resulted in the misguided application of results from studies of traumatic brain injury (TBI), stroke, Reye syndrome, and cerebral protection to patients suffering cardiopulmonary arrest. Second, within cardiopulmonary arrest etiologies and patient-relevant biologic factors such as genetic influences and comorbidities are lumped together. Factors influencing neurologic damage and recovery are clearly different depending on the cause (asphyxia, arrhythmia, hemorrhage, trauma, sepsis, etc.), age of the patient, interval between arrest and return of spontaneous circulation (ROSC), and effectiveness of cardiopulmonary resuscitation (CPR). Finally, the frustration encountered in dealing with the poor neurologic outcomes that often occur in this syndrome has reduced the enthusiasm for clinical studies involving novel treatments or those with proven efficacy for other indications.
Epidemiology
In the United States, cardiac arrest occurs in 8 to 20/100,000 children per year in the out-of-hospital setting and in 1.06 of every 1000 pediatric hospital admissions.6,9,10 Males have an increased frequency of cardiac arrest (~60 vs. 40% for females) but there are no sex differences in mortality.7 Half of children with cardiac arrest have underlying comorbidities.7,11
Asphyxia is the most prevalent cause of cardiac arrests and the principal cause of HIE in children.12,13 In asphyxial arrest, asystole or pulseless electrical activity is preceded and precipitated by a period of hypoxemic or anoxic perfusion.14 Hypoxia most commonly results from submersion accidents, airway obstruction, pulmonary aspiration, severe asthma or pneumonia, inhalation injury, or apnea syndromes.6,7,15 In contrast, ventricular fibrillation (VF) is the most frequent cause of out-of-hospital cardiopulmonary arrest in adults.16 In VF-induced cardiac arrest, respiration ceases shortly after loss of perfusion pressure. VF also occurs in children, but at an estimated incidence of less than 10% of pediatric victims of cardiac arrest overall, more commonly in the in-hospital setting.7
Cellular and Molecular Pathobiology
Mechanisms of Hypoxic-Ischemic Brain Injury
Cerebral neurons in culture can tolerate hours of extreme hypoxia. Although it takes about 160 minutes of exposure to an anoxic gas mixture for oxygen tension in the culture medium to reach 1 mm Hg, cortical neurons tolerate 1 to 3 additional hours with little histologic change.17 If 1 mmol/L sodium cyanide is used to simulate immediate anoxia, hippocampal neurons become swollen and vacuolated within 20 to 60 minutes and begin to disintegrate in 4 hours. Similarly, even 1 hour of complete global brain ischemia in monkeys is followed by electrophysiologic recovery of many neurons and significant recovery of some aspects of brain metabolism, such as protein synthesis.18 Although the time limit for consistently normal outcome after normothermic primary cardiac arrest is unknown, it is certainly closer to 5 to 10 minutes than 1 to 3 hours. Restoration of integrated brain function, that is, “neurologic recovery,” differs markedly from physiologic or metabolic brain recovery. In contrast to the relative cellular homogeneity in other organs, the functional specificity and interactions of neurons and glia in brain make patchy areas of cell death potentially devastating. This is evident in the neuropathology of dogs in persistent coma 1 week after a 10- to 15-minute cardiopulmonary arrest. Scattered neuronal death is evident, but the vast majority of neurons appear normal.19
Energy Failure
The brain depends on large amounts of substrate (glucose and lactate) because of its tremendous metabolic demands and paltry energy stores. Interruption of cerebral blood flow (CBF) results in loss of consciousness and electroencephalographic silence within seconds. Within 5 to 7 minutes, energy failure occurs, accompanied by disturbances of ion homeostasis in neurons and glial cells. Clinically this rapid depletion of brain high-energy phosphates has been demonstrated after hypoxia-ischemia in neonates using phosphorus magnetic resonance spectroscopy.20 Influx of sodium and water and efflux of potassium occur because the cells cannot maintain their energy-dependent electrochemical gradients. When the extracellular potassium concentration reaches 10 to 15 mmol/L, voltage-gated channels open and extracellular calcium influx occurs.21
If CBF remains inadequate and energy failure persists, calcium-mediated events such as phospholipase and protease activation can lead to irreversible injury and neuronal cell death. Cerebral acidosis occurs, and intracellular pH decreases from 7 to 6.4.22 If CBF is restored, however, recovery of basal cellular metabolism (ATP levels, protein synthesis, and oxygen consumption) and pH occurs. This has been shown in brain tissue samples and intact brain measurements after global ischemic insults that result in persistent vegetative coma.18
After anoxia or ischemia, the recovery of aerobic metabolism is essential for recovery, but is not in and of itself sufficient. The imbalance between aerobic and anaerobic metabolism and overdependence of neurons on lactate as a substrate23 must be restored. Despite global metabolic recovery, certain neurons progress to cell death. After restitution of CBF and oxidative metabolism after energy failure in the brain cellular and molecular dysfunction progresses and cells may die via immediate necrosis (complete energy failure), programmed cell death (apoptosis, autophagic stress, or delayed necrosis), or a spectrum of these processes.24–26 Brain magnetic resonance spectroscopy demonstrates early (during ischemia) and late (48 hours after reperfusion) depletion of high-energy phosphorous compounds and a corresponding lactate peak occurring in the face of normal vital signs, serum glucose, and arterial oxygen saturation after experimental hypoxia-ischemia.27,28
Selective Vulnerability
Certain neurons, such as those in the CA1 region of the hippocampus; basal ganglia; cerebral cortical layers III and V; portions of the amygdaloid nucleus; the cerebellar Purkinje cells; and in infants, periventricular white matter regions and some brainstem nuclei, are known to be especially vulnerable to hypoxia-ischemia.29,30 Five minutes of complete global brain ischemia produces cell death in these regions that begins to appear between 48 and 72 hours, without apparent histological damage in other brain areas.
Transient calcium accumulation occurs in all cells during ischemia, but secondary irreversible accumulation occurs in the selectively vulnerable zones many hours later.31 Electrophysiologic studies show that delayed neuronal death is preceded by neuronal hyperactivity. It is hypothesized that ischemic and early postischemic calcium accumulation leads to a complex sequence of derangements in cellular metabolism such as protease activation and oxygen-derived free radical formation.32 These conditions, in concert with excessive release of excitatory neurotransmitters (glutamate, glycine, and aspartate) in these areas, lead to excitotoxicity and cell death. These findings are supported by work in neuronal culture showing that calcium influx accompanies cell death in the presence of anoxia or supraphysiologic levels of excitatory amino acids such as glutamate,17,33 and that CA1 cells are the most sensitive to glutamate-mediated injury.34 Finally, delayed energy depletion, mitochondrial dysfunction, and infarction occur in concert, but are regionally distinct, suggesting that metabolic characteristics of brain regions affect recovery from ischemia.35,36
Of particular interest is that these intrinsically vulnerable cells do not have a unique vascular distribution.37 They represent neither vascular watersheds nor hypoperfused zones. Death of these neurons after a threshold ischemic insult occurs in a delayed fashion after reperfusion and thus may be preventable, at least in part, by treatment.
Cell Death Mechanisms
Cell death can occur by at least three distinct pathways, necrosis, apoptosis or autophagy.24,38 Cell death can also occur via a combination of one or all three of these pathways (see also Chapter 100). Necrosis, which is characterized by denaturing and coagulation of cellular proteins, is the basic pattern of pathological cell death that results from progressive reduction in the cellular content of adenosine triphosphate.39 Necrosis involves progressive derangements in energy and substrate metabolism that are followed by a series of morphologic alterations, including swelling of cells and organelles, subsurface cellular blebbing, amorphous deposits in mitochondria, condensation of nuclear chromatin, and finally, breaks in plasma and organellar membranes.39 Whereas necrotic cell death was traditionally felt to be entirely irreversible, recent studies showing that some degree of necrotic cell death responds to treatment after hypoxia-ischemia implicate “programmed necrosis,” also termed “necroptosis.”40
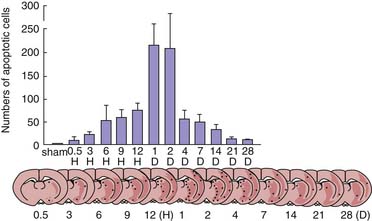
Cell death after hypoxic-ischemic insults can also occur by apoptosis.41 Development of apoptosis usually requires new protein synthesis and the activation of endonucleases. Two distinct types of characteristic cleavage of DNA have been described. The most well described, caspase-dependent apoptosis, involves cleavage by caspase activated deoxyribonuclease at linkage regions between nucleosomes to form fragments of double-stranded DNA.42 This produces a pattern of DNA cleavage observed on Southern blot analysis termed “DNA laddering.” In contrast, caspase-independent apoptosis results in large-scale DNA fragmentation induced by the mitochondrial flavoprotein apoptosis-inducing factor.43,44 The stimuli triggering apoptosis are complex and multifactorial, involving extracellular surface receptors, cell signaling pathways, protease cascades, and mitochondrial and other organelle dysfunction. Selective vulnerable cell death in brain regions such as the CA1 region of the hippocampus after transient global brain ischemia appears to occur by apoptosis.45 DNA extracted from the hippocampus of gerbils at 4 days after a threshold global ischemic insult exhibits a characteristic laddering pattern consistent with apoptotic cell death. Thus after a threshold ischemic insult, selective neuronal death in the highly vulnerably CA1 region of the hippocampus occurs by apoptosis. Thalamic-delayed neuronal death was caused by Fas-mediated apoptosis in a model of neonatal hypoxia-ischemia.46 Li et al.47 reported that apoptosis in the postischemic brain is not limited to scattered neuronal death in what have been traditionally deemed to be “selectively vulnerable regions,” but is seen even in penumbral regions around evolving cerebral infarcts (Figure 62-1). Finally, the proportion of apoptosis in the developing brain after ischemia appears to be a sex-dependent, because females but not males respond to antiapoptotic agents after neonatal hypoxia-ischemia.48
Recently, a new mode of cell death after brain ischemia has been implicated—autophagy. Autophagy is a homeostatic process that recycles cell resources during periods of nutrient stress.49 Autophagy is upregulated after experimental brain ischemia,50,51 which can be considered profound nutrient stress.52 Studies show that blocking autophagy after hypoxia-ischemia can be protective or detrimental.38,53,54 As such, autophagy’s role after acute brain injury is controversial, and it may depend on stage of brain development and sex of the patient, animal, or cell.52,55 Mouse pups lacking Atg7 (a necessary component for autophagy) or rat pups treated with an inhibitor of autophagy (3-methyladenine) are protected from focal hypoxia-ischemia.50,51
The proportion of neuronal cell death that occurs via apoptosis, necrosis, or autophagy after cerebral ischemia remains undetermined.56,57 Moreover, it remains possible that treatments inhibiting apoptosis may simply convert cell death to necrosis or autophagy.58,59 Although speculative, it is quite possible that after cardiopulmonary arrest and resuscitation, a continuum exists in neurons from recovery to necrosis60 that depends on the duration of the insult, the local milieu, and the given brain region.61–63
Nevertheless, in any given brain region, whether neuronal death is produced by necrosis, apoptosis, or autophagy, a highly complex series of events appears to be involved during the arrest and after ROSC. A theoretical scheme of the mechanisms involved is given in Figure 62-2, a scheme that remains remarkably contemporary despite being conceptualized almost two decades ago by Bellamy, Safar, and others.64 Importantly, several molecular and pharmacological interventions that interrupt apoptosis have been reported to improve outcome after cerebral ischemia, including “pediatric” models. Although studies showing improved functional recovery with treatments inhibiting apoptosis after cerebral ischemia suggest that some salvaged neurons regain their integrative properties, it is also possible that some of the delayed apoptosis represents appropriate pruning of neurons that have lost their targets. These processes, coupled with the need to restore highly integrated function, explain the unfortunate clinical scenario of the persistent vegetative state despite restoration of normal function in other organ systems. The relationship of these distinct forms of cell death to selective vulnerability of neurons in brain is beginning to emerge.
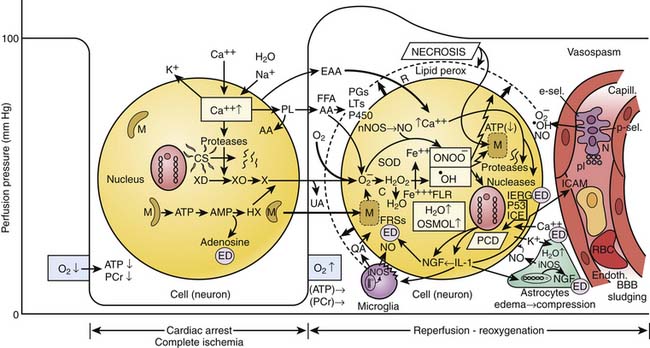
Figure 62–2 Death of cells after temporary ischemia. Diagram of complex, partially hypothesized biochemical cascades in neurons during and after cardiac arrest.64 Normally intracellular ([Ca2++]i) to extracellular ([Ca2++]e) calcium gradient is 1:10,000. Calcium regulators include calcium/magnesium adenosine triphosphatase, the endoplasmic reticulum (ER), mitochondria, and arachidonic acid (AA). With stimulation, different cell types respond with an increase in [Ca2++]i because of release of bound Ca2++ in the ER and influx of [Ca2++]e, or both. During complete ischemic anoxia (cardiac arrest) (left), the level of energy (phosphocreatinine [Pcr] and adenosine triphosphate [ATP]) decreases to near zero in all tissues at different rates, depending on stores of oxygen and substrate; it is fastest in the brain (~5 minutes) and slower in the heart and other vital organs. This energy loss causes membrane pump failure, which causes a shift of sodium (Na+) ions, water (H2O), and calcium ions (Ca2++) from the extracellular into the intracellular space (cytosolic edema), and potassium (K++) leakage from the intracellular into the extracellular space. Increase in [Ca2++]i activates phospholipase A2, which breaks down membrane phospholipids (PL) into free fatty acids (FFA), particularly AA. Increase in [Ca2++]i also activates proteolytic enzymes, such as calpain, which may disrupt the cytoskeleton (CS) and possibly the nucleus. In mitochondria (M) hydrolysis of ATP to adenosine monophosphate (AMP) leads to an accumulation of hypoxanthine (HX). Increased [Ca2++]i may enhance conversion of xanthine dehydrogenase (XD) to xanthine oxidase (XO), priming the neuron for the production of the oxygen free radical superoxide anion (O2) although this pathway is of questionable importance in neurons (X, xanthine; UA, uric acid). Excitatory amino acid neurotransmitters (EAA), particularly glutamate and aspartate, increase in extracellular fluid. Increased [EAA]e activates N-methyl-D-aspartate (NMDA) and non-NMDA receptors (R), thereby increasing calcium and sodium influx and mobilizing stores of [Ca2++]i. Increased extracellular potassium activates EAA receptors by membrane depolarization. Glycolysis during hypoxia results in anaerobic metabolism and lactic acidosis, until all glucose is used (in the brain, during anoxia after ~20 minutes). This lactic acidosis, plus inability to wash out CO2, results in mixed tissue acidosis that adversely influences neuronal viability. The net effect of acidosis on the cascades during and after ischemia is not clear. Mild acidosis may actually attenuate NMDA-mediated [Ca2++]i accumulation. Without reoxygenation, cells progress via first reversible, later irreversible structural damage, to necrosis at specific rates for different cell types. During reperfusion and reoxygenation (right), lactate and molecular breakdown products can create osmotic edema and rupture of organelles and mitochondria. Recovery of ATP and Pcr and of the ionic membrane pump may be hampered by hypoperfusion as a result of vasospasm, cell sludging, adhesion of neutrophils (granulocytes) (N), and capillary compression by swollen astrocytes, which also help to protect neurons by absorbing extracellular potassium. Capillary (blood-brain barrier [BBB]) leakage results in interstitial (vasogenic) edema. Increased concentrations of at least four oxyradical species that break down membranes and proteins, worsen the microcirculation, and possibly also damage the nucleus may be formed: Superoxide anion (O2.−) leading to hydroxyl radical (−OH) (via the iron-catalyzed Fe+++ → Fe++, Haber-Weiss/Fenton reaction); free lipid radicals (FLR) and peroxynitrite (OONO−). O2 may be formed from several sources: (1) directly from eicosanoid metabolism; (2) by the previously described XO system; (3) via quinone-mediated reactions within and outside the electron transport chain (from mitochondria [M]); and (4) by activation of NADPH-oxidase in accumulated neutrophils in the microvasculature or after diapedesis into tissue. Increased O2 leads to increased hydrogen peroxide (H2O2) production as a result of intracellular action of superoxide dismutase (SOD). [H2O2] is controlled by intracellular catalase. Increased O2 further leads to increased.OH, because of conversion of H2O2 to −OH via the Haber-Weiss/Fenton reaction, with iron liberated from mitochondria. This reaction is promoted by acidosis..OH and OONO− damage cellular lipids, proteins, and nucleic acids. Also, AA is metabolized by the cyclooxygenase pathways to prostaglandins (PGs) including thromboxane A2, or by the lipoxygenase pathway to produce leukotrienes (LTs); and by the cytochrome P-450 pathway. These products can act as neurotransmitters and signal transducers in neuronal and glial cells and can activate thrombotic and inflammatory pathways in the microcirculation. Inflammatory reactions after ischemia have been shown to occur in extracerebral organs, focal brain ischemia, or brain trauma; to date, they have not been demonstrated after temporary complete global brain ischemia. Neuronal injury can signal interleukin-1 and other cytokines to be produced and trigger endogenous activation of microglia, with additional injury (QA, quinolinic acid). In addition, tissue or endothelial injury—particularly associated with necrosis—can signal the endothelium to produce adhesion molecules (intercellular [ICAM], e-selectin [e-sel], p-selectin [p-sel]), cytokines, chemokines, and other mediators, triggering local involvement of systemic inflammatory cells in an interaction between blood and damaged tissue. Reoxygenation restores ATP through oxidative phosphorylation, which may result in massive uptake of [Ca2++]i into mitochondria, which are swollen from increased osmolality. Thus mitochondria loaded with bound [Ca2++] may self-destruct by rupturing and releasing additional free radicals. Increased [Ca2++]i by itself and by triggering free radical reactions may result in lipid peroxidation, leaky membranes, and cell death. Neuronal damage can be caused, in part, by increased [EAA]e (excitotoxicity). During reperfusion, [Ca2+]i and increased [EAAs]c normalize. Their contribution to ultimate death of neurons is more likely through the cascades they have triggered during ischemia. During ischemia and subsequent reperfusion, loading of cells and calcium maldistribution in cells are believed to be the key trigger common to the development of cell death. This calcium loading signals a wide variety of pathological processes. Proteases, lipases, and nucleases are activated, which may contribute to activation of genes or gene products (i.e., interleukin-converting enzyme, [ICE] or P53) critical to the development of programmed cell death (PCD, i.e., apoptosis, autophagy or necroptosis), or inactivation of genes or gene products normally inhibiting this process. Activation of nNOS by calcium can lead to production of NO., which can combine with superoxide to generate peroxynitrite (OONO−). OONO− and −OH both can lead to DNA injury and PCD, or protein and membrane peroxidation and necrosis, respectively. Nerve growth factor (NGF), nuclear immediate early response genes (IERG) such as heat-shock protein, free radical scavengers (FRSs), adenosine, and other endogenous defenses (ED) may modulate the damage.
(Courtesy P. Safar, MD, and P. Kochanek, MD, with input from N. Bircher, MD, and J. Severinghaus, MD.)
Reperfusion Injury
Reoxygenation and reperfusion are essential to recovery of any organ after ischemia. Experimental evidence suggests, however, that certain aspects of reperfusion result in tissue injury.65,66 Reperfusion injury is a complex series of interactions between parenchyma and microcirculatory elements, resulting in detrimental effects that negate some fraction of the benefits of reperfusion. The magnitude of reperfusion injury varies with the organ in question, the duration and type of hypoxic-ischemic insult, as well as the timing, duration and magnitude of reperfusion.67,68
In many organs and in the brain after focal insults, progressive microcirculatory failure is thought to be an important aspect of reperfusion injury.69 However, as suggested by the nonspecific vascular distribution of selectively vulnerable neurons, the brain may display a second unique setting for the evolution of reperfusion injury, the evolution of selectively vulnerable cell death. Four key mechanisms hypothesized to be important to reperfusion injury in the brain include: (1) excitotoxicity and calcium accumulation, (2) protease activation, (3) oxygen radical formation, and (4) membrane phospholipid hydrolysis and mediator formation.
Excitotoxicity and Calcium Accumulation
Glutamate and aspartate are the major excitatory amino acid neurotransmitters in the mammalian central nervous system, but both also have neurotoxic properties. Pioneering studies by Rothman et al.70 demonstrated in vitro that hypoxia-induced neuronal death is mediated by synaptic activity. Inhibition of synaptic glutamate release or blockade of glutamate receptors prevented hypoxia-induced neuronal injury. Glutamate is the major neurotransmitter in the selectively vulnerable zones and accumulates extracellularly at supraphysiologic levels in these regions after hypoxic or ischemic insults.31
Glutamate is released at the presynaptic terminal in response to neuronal stimulation and acts by binding to postsynaptic dendritic receptors. Two main classes of excitatory neurotransmitter receptors have been identified. One class consists of the ligand-gated ion channels (“ionotropic” receptors) and includes NMDA (N-methyl-D-aspartate), AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) or quisqualate, and kainate receptor subtypes. Toxicity caused by NMDA receptor activation is usually rapid, whereas AMPA or kainate receptor-mediated cell death is somewhat slower to develop.71 The other class of excitatory neurotransmitter receptors includes the “metabotropic” receptors. These receptors are coupled with G proteins and modulate intracellular second messengers such as calcium, cyclic nucleotides, and inositol triphosphate.72 When activated, the ionotropic glutamate receptors open sodium channels and may also have an important role in initiation and propagation of membrane depolarization and spreading depression.73 With ionotropic receptor activation, rapid excitatory amino acid-mediated calcium accumulation occurs. In the face of ischemia this calcium accumulation is exacerbated by cellular energy failure, that disables the Na+/K+-ATPase membrane pump and results in further calcium accumulation.31 Reestablishment of the energy supply can reverse these changes. Delayed glutamate-related neuronal injury is most likely the result of activation of ionotropic receptors and subsequent calcium influx. Calcium influx causes death of neurons in culture under anoxic conditions or in the presence of glutamate.31 The intracellular accumulation of calcium: (1) activates proteases, lipases, and endonucleases resulting in the breakdown of membrane phospholipids; (2) activates neuronal nitric oxide synthase (nNOS), resulting in nitric oxide (NO) production and, in the presence of superoxide, peroxynitrite formation; (3) damages mitochondria; (4) disrupts nucleic acid sequences; and (5) ultimately mediates cell death via necrosis, apoptosis or autophagy (see Figure 62-2). The disturbance of the finely regulated intracellular calcium homeostasis is now recognized as a possible final common pathway of neuronal death.25,31,64,72 NMDA receptor activation has also been shown to stimulate superoxide anion production, which may contribute to cellular injury.74 The NMDA and AMPA-receptor subtypes have been suggested to play key roles in ischemic brain injury. The potential implications of these mechanisms in regard to therapeutic manipulation of glutamate receptors, as well as calcium and sodium channels, are discussed later.
Protease Activation
One of the candidates for a critical role in neuronal injury as a result of increases in intracellular calcium concentration is protease activation. Protease activation plays a central role in mediating both necrosis and apoptosis. With regard to necrosis, numerous calcium-dependent enzymes can become activated during ischemia and produce important structural injury to neurons. One class of calcium-dependent proteases, calpains, are cytosolic cysteine proteases that have a homeostatic role in cell-cycle regulation, differentiation, cell migration, and signal transduction.75 After injury, calpain-mediated proteolysis of cytoskeletal proteins as well as activate protein kinase C and phospholipases can occur.75 Calpains also hydrolyze the major plasma membrane sodium/calcium exchanger in neurons during excitotoxicity, allowing enormous secondary influx of calcium into the cell following NMDA receptor activation and creating a positive feedback environment.76 In a model of oxygen-glucose deprivation in neuronal cell culture that mimics transient ischemia, calpains induced mitochondrial release of apoptosis-inducing factor, that translocates to the nucleus to initiate caspase-independent apoptosis.77
With regard to apoptosis, the caspase family of cysteine proteases plays an important role after cerebral ischemia, and may have a more prominent role in the developing versus mature mammalian brain.78,79 After unilateral hypoxic-ischemic brain injury, neonatal rats had increased cytochrome c release and caspase-3 activation versus juvenile and adult rats.55 Regulation of apoptotic machinery also appears to be sex dependent after neonatal hypoxic-ischemic brain injury.80 Comparatively, female rats had more caspase-mediated apoptosis while male rats had more caspase-independent, apoptosis-inducing factor-mediated apoptosis.
Oxygen Radical Formation
Toxic oxygen radical species produced during postischemic reperfusion have been implicated as important contributors to reperfusion injury and delayed cell death.66 The primary species of interest include superoxide anion, hydrogen peroxide, hydroxyl radical, and the reactive nitrogen species peroxynitrite.
The potential sources of oxygen radicals are many. Superoxide anion is produced by the electron transport chain during normal mitochondrial respiration. Mitochondrial dysfunction, as may occur under conditions of ischemia, produces increased generation of free radicals that may extend beyond the capacity of local endogenous antioxidants leading to oxidative stress. Metabolism of arachidonic acid via the cyclooxygenase pathway to form prostaglandins also produces superoxide anion as an enzymatic byproduct, and this also occurs to a lesser extent in the lipoxygenase and cytochrome P-450 pathways.66,81 Accumulated neutrophils also can contribute superoxide anion via neutrophil reduced nicotinamide adenine dinucleotide (NADPH)-oxidase. Yet another potential oxygen radical source is xanthine oxidase (XO)-xanthine dehydrogenase (XD). Energy depletion is associated with conversion of ATP to adenosine diphosphate (ADP) and eventually to hypoxanthine. This ischemia- or anoxia-induced energy deprivation also causes conversion of XD to XO via calcium-activated proteases. In the presence of hypoxanthine and oxygen, that become available during reperfusion, XO produces superoxide anion. These XO-mediated reactions may occur in cerebral vascular endothelial cells, that are rich in XO.66 However, the importance of XO in contribution to the generation of free radicals in human beings has been challenged.82,83 Autooxidation of circulating catecholamines or of neurotransmitter catecholamines may represent another potential source of oxygen radicals.
Another possible contributor to free radical generation is delocalized iron. Iron is normally transported in the blood tightly bound to transferrin and stored inside the cell bound to ferritin. In ischemic conditions with accompanying acidosis, however, iron may be displaced from its normal binding sites and can catalyze reactions that promote oxygen radical formation.84,85 Most commonly implicated is the Haber-Weiss/Fenton reaction, whereby the very potent hydroxyl radical is produced from superoxide anion and hydrogen peroxide in the presence of free iron. The iron-chelator, deferoxamine, has been shown to reduce neurologic injury after experimental neonatal hypoxia-ischemia.86,87 Finally, NO is another free radical that contributes to both nitrosative and oxidative stress. NO increases during ischemia via increased NMDA receptor stimulation, mediated by release of excitatory amino acids and subsequent calcium-mediated activation of nNOS.88 NO in the presence of superoxide produces peroxynitrite.89 Free radicals have also been associated directly with an increase release of excitatory amino acids and vice versa.74,90 Not only do they participate in each other’s release and formation, but they also may act synergistically in causing tissue damage.
The brain may be particularly vulnerable to free radical injury for several reasons. One is the high concentration of polyunsaturated fatty acids, especially arachidonic acid. Release of free fatty acids (FFA) occurs throughout ischemia. On exposure to oxygen radical species, these FFA are vulnerable to autocatalytic lipid peroxidation.91 Cerebrospinal fluid (CSF) has low concentrations of iron-binding proteins; therefore, iron released from injured neurons or glia is likely to contribute to these peroxidation reactions. Byproducts of these reactions, for example, malondialdehyde and conjugated dienes, have been used as markers of the extent of lipid peroxidation after brain injury (e.g., the thiobarbiturate assay). Lipid peroxides accumulate in the selectively vulnerable zones during reperfusion after transient forebrain ischemia.84,92 The peroxides do not accumulate during the ischemic period itself or in areas that are not reperfused and thus are implicated in reperfusion injury.93
Investigators have also detected oxidative damage to brain proteins after reperfusion.93 Pyruvate dehydrogenase, a key mitochondrial matrix enzyme that converts pyruvate into acetyl coenzyme A, undergoes oxidative protein modification after ischemia that impairs enzyme activity, possibly contributing to neuronal cell death.94 However, others have argued that oxidized proteins detected in previous studies were oxidized during sampling and not as a result of ischemia.91 Other investigators were also unable to detect oxidized nuclear material or mitochondrial DNA during cerebral ischemia.95 These results were expected because of the compact structure of DNA and the presence of histones, which provide increased resistance to oxidative damage. However, there are two problems with these findings: (1) damage caused by oxidative stress can occur in the absence of lipid peroxidation96 and (2) work investigating apoptosis suggests that oxidation of regulatory proteins of transcription and translation of DNA may be an important intermediate step in propagation of neuronal cell death.97,98 Application of contemporary techniques to quantify oxidative stress should help clarify this issue,99 and may assist in the evaluation of therapies to prevent oxidative stress-mediated brain damage produced during ischemia and reperfusion.
Developmental and sex differences exist in terms of the degree of oxidative stress and amount and function of antioxidant enzymes after brain injury. In mice, glutathione and catalase activity in brain is higher in adult female versus male mice, and discrepancies become more exaggerated with age.100 Furthermore, neurons from male rats have less capacity to replenish glutathione levels after cytotoxic stress in vitro, and in vivo after asphyxial cardiac arrest.101 A similar pattern occurs in adult patients after severe TBI with males demonstrating increased lipid peroxidation products in CSF compared with females.102
Membrane Phospholipid Hydrolysis and Mediator Formation
Membrane phospholipids modulate signaling cascades, affecting the development, differentiation, function, and repair of the nervous system, functions that become dysregulated with ischemia and oxidative stress.103 FFAs are released from neuronal membranes during ischemia, and the amount of FFA released are proportional to the duration of ischemia. FFA release continues to change in proportion to duration of ischemia after the completion of energy failure.104 FFA are released by two distinct but related processes. First, phosphatidyl inositol is hydrolyzed by phospholipase C with the production of diacylglycerol (DAG) and inositol phosphates.105,106 Phospholipase C-mediated hydrolysis begins during the initial moments of the ischemic insult and is related to neurotransmitter receptor stimulation. DAG is then hydrolyzed by lipases to FFA, predominantly arachidonic and stearic acid. Second, other brain glycerophospholipids are hydrolyzed by phospholipase A2, which is activated by increases in intracellular calcium concentration. FFA release and metabolism is not a generalized process in the neuronal membrane, but is concentrated in the synaptic regions and is thus related to excitotoxicity.107
The FFAs released then have potential detrimental effects during the postischemic period by three mechanisms: (1) arachidonic acid metabolism via the cyclooxygenase pathway contributes to oxygen radical production during reperfusion81; (2) FFA and DAG directly increase membrane fluidity, inhibit ATPases, increase neurotransmitter release, and uncouple oxidative phosphorylation; and (3) enzymatic oxidation of arachidonic acid during reperfusion by cyclooxygenase, lipoxygenase, or cytochrome P-450 produces a large number of bioactive lipids, including prostaglandins, thromboxanes, leukotrienes, and hydroxy fatty acids, many of which have detrimental effects (see Figure 62-2).
Endogenous Defenses
The heat-shock proteins are one family of candidate neuroprotectants that are highly conserved among biological species and are induced in cells after a variety of stimuli. Thermal stress is the classic example; however, any insult that damages protein structure, including ischemic108 and TBI,109 can produce a heat shock protein response. Simon et al.108 showed that after global ischemia the 72-kDa heat shock protein (Hsp72) is temporally expressed in a pattern that mirrors the pattern of selective vulnerability in the model, seen first in the CA1 region of the hippocampus, followed by CA3, cortex, and thalamus, and finally in the dentate granule cells. Hsp72 is also induced in both gray and white matter of piglets after mild and severe hypoxia.110 The heat shock proteins have generated major interest as potential neuroprotectants because their prior induction by a sublethal stress can afford protection from subsequent injury. Transient whole-body hyperthermia reduces subsequent ischemic brain injury in both adult111 and neonatal rats.112 Furthermore, exogenous Hsp72 reduces glutamate toxicity in neuronal cell cultures.113 Importantly, overexpression of Hsp72 reduces ischemic damage and apoptosis after experimental stroke and global ischemia in vivo.114–116
Another potential mechanism for endogenous neuroprotection is the upregulation of genes that inhibit apoptosis and augment neurogenesis. The mammalian gene bcl-2, a proto-oncogene, can block apoptosis97 and perhaps necrosis as well.117 The bcl-2 gene is expressed in neurons surviving both focal and global ischemia118,119 and is reduced in degenerating neurons after cardiopulmonary arrest in rats.120 Viral transfection of bcl-2 reduces infarction after focal ischemia,121 and upregulation of bcl-2 via ceramide administered 30 minutes after hypoxia-ischemia reduced the number of cells with DNA damage in immature rat brain.122 Forced overexpression of the bcl-2 family member bcl-xL also reduces tissue damage after focal cerebral ischemia in adult rats.123 After TBI in infants and children, CSF levels of bcl-2 are increased in patients who survive compared with those who die.124 Finally, bcl-2 overexpression promotes neurogenesis in adult mice with and without ischemic injury.125
Adenosine is an endogenous biochemical mediator that may serve a protective role after cerebral ischemia, particularly early after injury. Adenosine is increased in brain tissue after experimental ischemia,126 and in response to hypoxia,127 hypotension,128 and hypoglycemia.129 The release of adenosine after ischemia could afford neuroprotection by a combination of several mechanisms. When bound to A2-receptors, adenosine is a potent cerebrovasodilator and inhibits platelet activation and neutrophil function.130 Bound to A1-receptors, adenosine reduces neuronal metabolism and excitatory amino acid release and stabilizes postsynaptic membranes.130 Thus the beneficial effects of adenosine after cerebral ischemia include improved regional blood flow, reduced local oxygen demand, attenuation of both excitotoxicity and calcium accumulation, and antiinflammatory and rheologic effects. Finally, adenosine agonists have been shown to improve survival of selectively vulnerable neurons after ischemia in many studies (reviewed in Rudolphi et al.131).
Clinical Pathophysiology
Cerebral Blood Flow and Metabolism After Resuscitation
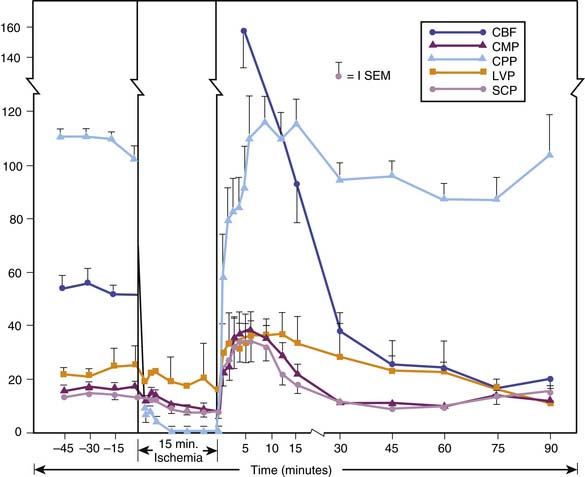
The pioneering studies in which global CBF and cerebral metabolic rate for oxygen (CMRO2) were measured in animal models of global ischemia or cardiac arrest focused on the early post-resuscitation period. In their classic study, Snyder et al.132 showed that after 15 minutes of global brain ischemia in dogs, CBF transiently increased to levels well above baseline (Figure 62-3). Then, after 15 to 30 minutes, CBF progressively decreased to a level below normal for the remainder of the monitoring period (90 minutes). This pattern of early transient postischemic hyperemia and subsequent delayed postischemic hypoperfusion has been observed in many global cerebral ischemia models, including both VF and asphyxial arrest.133,134 The level of hyperemia and subsequent hypoperfusion vary in relation to the duration of the insult.135 Although these phases of increased and decreased CBF characterize the net global effect, regional CBF is often inhomogeneous, particularly during postischemic hypoperfusion, when areas of decreased and increased perfusion may coexist.136
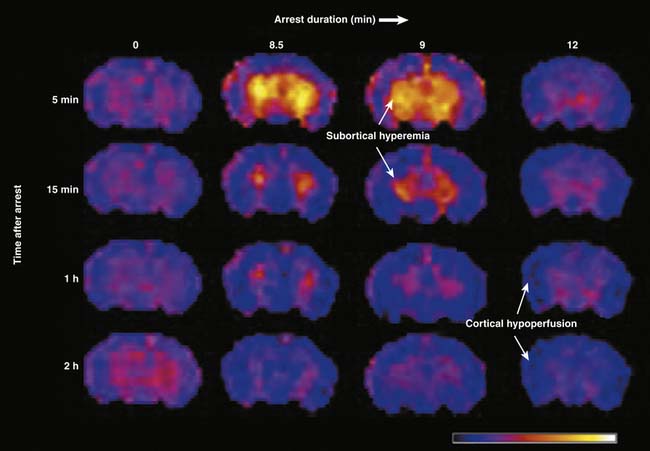
Recently, the heterogeneous and duration dependent nature of post-arrest CBF was characterized using contemporary imaging techniques allowing for regional assessment and a clinically relevant model of pediatric asphyxial cardiac arrest.137 Using arterial spin labeling magnetic resonance imaging, CBF was measured for the first 3 hours after 8.5, 9, or 12 minutes of asphyxial cardiac arrest in postnatal day 17 rats—approximating a 1- to 4-year-old human in terms of brain development (see also Chapter 57). Although the pattern of early global hyperemia followed by hypoperfusion similar to that observed by Snyder et al.132 was seen after asphyxial arrests lasting 8.5 and 9 minutes, a pattern of global and persistent hypoperfusion was observed after a 12-minute arrest (Figure 62-4). Remarkably, CBF disturbances were also found to be region-dependent after asphyxial arrests lasting 8.5 and 9 minutes, with subcortical hyperemia but cortical hypoperfusion. After a 12-minute asphyxial arrest hypoperfusion was observed in both cortical and subcortical regions, and CBF was pressure passive, perhaps indicating loss of autoregulation after a prolonged arrest.
In patients with good outcomes, global CBF recovers over the subsequent 24 to 72 hours and CO2 reactivity remains intact. Patients who do not regain consciousness or progress to brain death develop absolute or relative CBF hyperemia with impaired CO2 reactivity.138,139 A theoretical scheme of postarrest global CBF and its relation to neurologic outcome is presented in Figure 62-5. Most studies in experimental animal models of asphyxial arrest suggest a similar pattern of CBF and CMRO2 to that observed after VF cardiac arrest and global ischemia in the early post-resuscitation period in humans.134,138 However, there are some exceptions.140 Results from clinical studies of pediatric asphyxial arrest are scarce and somewhat conflicting with regard to the prognostic implications of high or low values of postarrest CBF based on a single measurement; however, loss of CO2 reactivity appears to be associated with poor outcome in all studies. In studies of children between 24 and 48 hours after near-drowning, Ashwal et al.141 observed low CBF in the seven nonsurvivors and no relationship between CBF and PaCO2 in these patients—again suggesting loss of CBF reactivity to changes in PaCO2. In this study, hyperemia was not routinely observed in either survivors in a persistent vegetative state or children who died, but only a single CBF measurement was made in these patients. Beyda142 obtained serial measurements of postarrest 133Xenon in a series of children who suffered asphyxial arrest from submersion accidents. Children with good neurologic outcomes had slightly decreased CBF values at 12 hours that increased to normal during the subsequent 24 to 60 hours. In these children, CBF reactivity to CO2 was intact. Children with eventual vegetative outcome or brain death exhibited hyperemia with loss or attenuation of CO2 reactivity. This hyperemia progressed to low or normal flow over the following 12 to 72 hours in children with vegetative outcome and progressed to low and then no flow with the development of brain death.
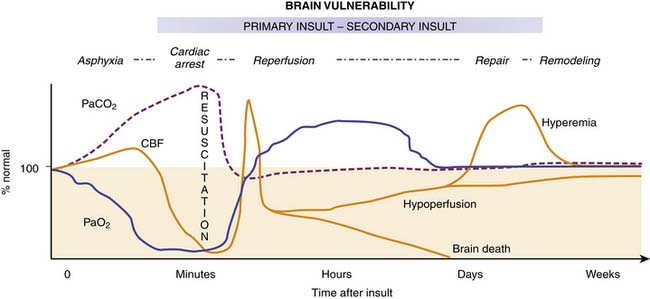
Figure 62–5 Hypothetical diagram illustrating the patterns of global cerebral blood flow (CBF) during and after cardiopulmonary arrest of moderate duration in humans. Immediately after resuscitation, “early postischemic hyperemia” occurs for about 15 minutes in subcortical brain regions. This is followed by patchy multifocal “delayed postischemic hypoperfusion” in cortical brain regions lasting from a few hours to days. Progressive return of CBF to normal is seen in patients with intact neurological outcome. In contrast, “delayed postischemic hyperemia” can be observed hours to days postarrest in patients with more severe insults.139,142 This delayed hyperemia appears to be associated with disabled or vegetative outcome (where CBF gradually decreases to near normal or below normal) or brain death (where CBF decreases to no flow). It is unclear, however, if all patients with vegetative outcome or eventual brain death develop delayed hyperemia.
Brain metabolism, as assessed by CMRO2, is reduced during the early postischemic period and then progressively recovers to a level that varies, depending on the model used and on the duration of ischemia.135,143 In some models, including VF arrest in dogs, significant recovery of CMRO2 may occur during the first few hours, despite persistent postischemic hypoperfusion—creating the potential for a secondary ischemic insult during reperfusion. Whether this increase in CMRO2 represents appropriate synaptic activity, seizures, or basal metabolism is not certain. In other models, global CBF and CMRO2 are matched during the first few hours after ischemia.
It must be recognized that the measurements of metabolism in the studies have traditionally been CMRO2. However, Bergsneider et al144 used positron emission tomography in patients after TBI and reported the occurrence of delayed hyperglycolysis in some comatose patients, suggesting a shift to anaerobic metabolism in injured brain in some patients. Using microdialysis in a porcine model of cardiac arrest, increased lactate/pyruvate ratios were found during arrest, and again in a delayed fashion especially if kept normothermic versus hypothermic.145 This same group found increased lactate/pyruvate ratios and glutamate using microdialysis in adult survivors after cardiac arrest, all of whom were treated with hypothermia.146 Prolonged increases in brain lactate detected using proton magnetic resonance spectroscopy after global hypoxia-ischemia in children have also been reported.28,147,148 Oxidative stress decreases the function of the pyruvate dehydrogenase complex, a key enzyme in oxidative metabolism, possibly contributing to the shift to anaerobic metabolism.149 Although routine monitoring of CBF or CMR has not been applied extensively to the post-arrest setting in children, their routine assessment using contemporary methods would likely lead to an improved understanding of pathophysiology.
Drugs such as nimodipine can increase CBF during the early postischemic hypoperfusion phase after global cerebral ischemia. CMRO2 recovery generally is not increased by treatment.150 Although nimodipine has been shown to be beneficial in patients after subarachnoid hemorrhage,151 but not ischemic stroke,152 the testing of strategies targeting early post-arrest hypoperfusion deserves further study. Safar et al.153 have reported that a multifaceted treatment strategy to increase CBF (“flow promotion”) and reduce CMR or CMRO2 early after VF in dogs improved outcome. This was accomplished by use of cardiopulmonary bypass (CPB), mild hypothermia, hemodilution, and transient hypertension.
Histopathology of Hypoxic-Ischemic Encephalopathy
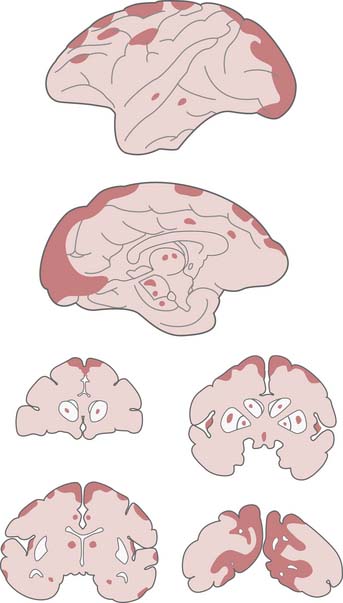
Ischemic neuronal change (Figure 62-6), as first described by Sommer in 1880 and later by Spielmeyer, involves a progression from extensive cellular microvacuolation to a cell that resembles a naked shrunken nucleus.18 As described by Brierley et al.,37 “this type of neuronal damage is neither ubiquitous nor randomly distributed but is found in regions which exhibit selective vulnerability to hypoxic stress.” As discussed previously, death of selectively vulnerable neurons (e.g., hippocampal CA1) cannot be explained by vascular distribution. Remarkably, these clinical descriptions of cell shrinkage were consistent with apoptosis rather than necrosis. However, the connection between selective vulnerability and apoptosis was made 100 years later.45 Neuronal death after cardiopulmonary arrest is seen not only in the selectively vulnerable neurons but also as a subtle histopathological finding in the arterial boundary zones. These neurons (not otherwise selectively vulnerable) are in the most poorly perfused areas during or after resuscitation.34 Neuronal death in the arterial boundary zones was elegantly described by Nemoto et al. in a monkey model of 16 minutes of complete global brain ischemia followed by 7 days of intensive care.154 Maximal damage appeared to be in the classically described selectively vulnerable zones, but neuronal death was also observed in the most distal distribution of the posterior cerebral artery and in the watershed zones of the anterior and middle cerebral arteries (Figure 62-7). With sufficient injury in the arterial boundary zone, more severe findings such as microinfarction or laminar necrosis can be seen.19,154 As previously discussed, even in stroke, neuronal death in an ischemic penumbra can occur either by necrosis or apoptosis. Thus it appears that there may be a continuum between apoptosis and necrosis that may depend on a large number of factors, such as duration of the insult and brain region in question.60 Whereas apoptosis in a given area often affects only a select percentage of neurons, infarction affects all neurons and glia. Obviously, if the arrest time is long or if the postischemic conditions are sufficiently poor, infarction of the entire brain can occur.
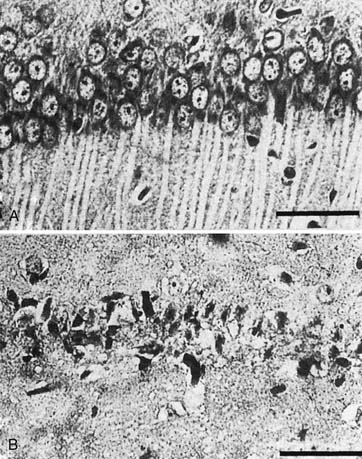
Figure 62–6 Light micrograph of dorsal hippocampus in gerbils 7 days after 5 minutes of sham ischemia (A) or global ischemia by carotid occlusion (B). Ischemic neuronal change is seen with CA1 neurons appearing as dark, shrunken nuclei without cytoplasm (B) by contrast with the normal-appearing nonischemic neurons (A). Investigations by Nitatori et al.45 indicate that neuronal death in selective areas, after threshold ischemia insults, occurs via apoptosis.
(From Kuroiwa T, Bonnekoh P, Hossmann KA: Therapeutic window of CA1 neuronal damage defined by an ultrashort-acting barbiturate after brain ischemia in gerbils, Stroke 21:1489, 1990.)
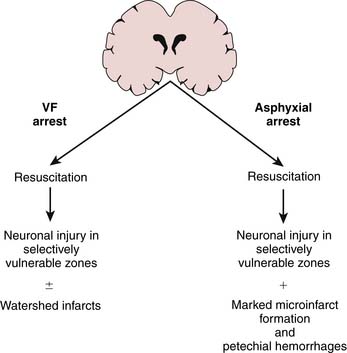
Vaagenes studied neuropathology after primary VF arrest of 10 minutes in dogs.19 Despite vegetative outcome at 96 hours, only scattered ischemic neuronal changes in the selectively vulnerable neurons and to a much lesser extent in the vascular watersheds were observed. Microinfarct formation was seen in only 5 of 18 dogs, suggesting that patchy ischemic neuronal change is sufficient for vegetative outcome. They then compared this 10-minute VF arrest with an asphyxial episode (airway occlusion) resulting in cardiac arrest with 7 minutes of no flow. Related either to differences in the initial insult or to postischemic events, asphyxial arrest resulted not only in ischemic neuronal change in the selectively vulnerable regions but also in marked microinfarct formation (30 of 32 dogs) and scattered petechial hemorrhages. This more severe histological injury was seen despite significantly easier ROSC in the asphyxial arrest group (Figure 62-8). In addition, unlike VF arrest, asphyxial arrest caused some ischemic neuronal changes even after no flow of only 2 minutes. These findings may explain the poor outcome generally observed in cardiopulmonary arrest in children (usually asphyxial arrest) compared with that in adult series (VF arrest). After asphyxial cardiac arrest that results in long-term survival in both adult and pediatric-aged animals,155–157 the pattern of neuronal death produced is similar to that reported in human studies158 including that of the young victim of asphyxial cardiac arrest, Karen Ann Quinlan,159 in which a predilection for basal ganglia injury resulting in a persistent vegetative state is observed.
Clinical Outcome After Pediatric Cardiopulmonary Arrest
Compared with results in adult series, survival and neurologic outcome after cardiopulmonary arrest in infants and children are remarkably poor.6 Most published data examining out-of-hospital cardiopulmonary arrest in adults reveal about 11% to 12% “good” neurologic outcome at 6 months after arrest, with good outcome generally defined as the ability to function independently.160 One review summarizing the results from 44 studies totaling 3094 pediatric patients after cardiopulmonary arrest showed favorable outcome at hospital discharge (generally defined as nonvegetative) in only about 6% of patients.13 In addition, after asphyxial arrest in children, clinical outcomes somewhere between vegetative and good, such as moderate disability or severe disability, are uncommon.141,161 Survival to hospital discharge after out-of-hospital arrest ranges from 8% to 25% and after in-patient arrest is 24% to 51% in several studies, with an overall survival of 13%.7,9,10,13,15 However, if the child had a pulse by hospital arrival after out-of-hospital cardiac arrest, survival increases to 38%.7 The most common cause of death is neurologic injury for out-of-hospital arrests and cardiovascular failure for in-hospital arrests.7 Good neurologic outcome in pediatric patients surviving cardiac arrest is often overestimated using traditional categorical outcome measures such as the Glasgow Outcome Score and Pediatric Cerebral Performance Category Scale.162–164 Furthermore, contemporary longer-term outcome studies evaluating overall functional status, neuropsychological function, and quality of life do not exist. One observational study focusing on late improvements in functional outcome noted improved mobility in 22% of children after cardiac arrest versus 66% of children after TBI two years after the initial event.165
This high mortality and poor outcome after cardiorespiratory arrest in children generally represents out-of-hospital or unwitnessed full arrests. Recovery is much better in children who had witnessed arrests, cold water submersion, or isolated respiratory arrest, after which intact survival rates as high as 44% to 75% have been reported.7,12,166–168 Nevertheless, these clinical data seem to bear out the severe neuropathology observed in asphyxia-induced arrest in animal models because asphyxial arrest is the most common mode of arrest in all of the clinical pediatric series.12,13,162,166–168
Such factors as initial pH, number of epinephrine doses, and arrest duration have been examined in an attempt to predict outcome from cardiopulmonary arrest. Although sometimes predictive, this information can be misleading. For example, the time delay before analysis of the first blood sample can vary, as can estimates of arrest duration. With asphyxial arrest, even controlled experimental animal studies show that the time from asphyxia until cardiac arrest varies considerably. Currently, the most powerful individual predictor of neurologic outcome after cardiorespiratory arrest is the neurologic examination or one of several scoring systems based on selected aspects of the neurologic examination. In 1985, Levy et al.169 applied multivariate analysis to variables from the neurologic examination in a series of 210 adults in coma studied for at least 6 hours after a hypoxic-ischemic insult. None of the 15 patients who were in a vegetative state at 1 month regained independent function. In this retrospective study, the combination of scores is from 2 weeks. The oculocephalic response and the 3-day motor response were able to identify all patients who gained independent function. This was true even for the 2 of 57 patients who remained in eyes-closed coma for 3 days and then ultimately regained independent function. Edgren et al.170 applied the Glasgow Coma Score (GCS) to assess outcome in 262 adult comatose survivors of cardiac arrest. GCS less than 6 at 3 days post-arrest predicted poor outcome (at 6 months) with no false-negative results. In a similar study of 216 adults after out-of-hospital cardiac arrest, Mullie et al.171 found that a best GCS of less than 4 as early as 2 days post-arrest predicted death or vegetative outcome in all but one patient. The one exception progressed from vegetative state to severe disability. The addition of hypothermia as a post-resuscitative therapeutic option may delay attempts at early prognostication.172
Although a few patients survive relatively intact despite poor prognosis they likely represent cases of asphyxia without cardiac arrest, coma of an origin other than asphyxial arrest, unrecognized hypothermia, or an associated overdose of a cerebral protective drug. Two prospective trials examining predictors of outcome have been reported after cardiopulmonary arrest in pediatric patients. Robertson et al. in a series of 27 pediatric patients with hypoxic-ischemic encephalopathy found that a GCS of 3 and nonreactive pupils were predictive of adverse outcome.162 Consistent with asphyxia as the predominant cause of cardiac arrest in infants and children, Sirbaugh et al. found that the only factor that was positively associated with ROSC was endotracheal intubation.173 This would not support abandonment of the practice of ventilating infants and children found pulseless and apneic, and rather, suggests that it is crucial in this patient population.174,175 Indeed a recent, prospective multicenter study done in Japan comparing conventional versus chest-compression only CPR for children after out-of-hospital cardiac arrest strongly supports this view.176 In children requiring resuscitation from non-cardiac causes, conventional CPR resulted in favorable neurologic outcome in 45 of 624 children compared with “hands-only” CPR where favorable neurologic outcome was seen in only 6 of 380 (7.2 vs. 1.6%; odds ratio, 5.54; confidence interval, 2.52 to 16.99). Importantly, in children requiring resuscitation from cardiac causes, favorable neurologic outcome was not different between conventional CPR and compression only CPR, implying that unlike in adults, conventional CPR does not negatively influence outcome in children suffering arrests of cardiac origin.
The electroencephalogram (EEG) can also provide prognostic information for patients after cardiac arrest. Scollo-Lavizzari and Bassetti177 retrospectively examined the relation between first post-arrest EEG and clinical outcome in 408 cases. A 5-grade classification was used to categorize EEGs. Although permanent severe neurologic damage was observed in some patients with grade I EEG, none of the 208 patients with grade IV or V EEGs had a good neurologic recovery. A recent single-center retrospective case series in children showed that after a 5-grade EEG score was collapsed into three categories, the score was associated with a positive predictive value of 90% and negative predictive value of 91% in determining poor outcome at hospital discharge.178 Detailed quantitative EEG may also have a role in the prediction of neurologic outcome after cardiac arrest (see also Chapter 34).179
Adjunctive prognostic information also can be derived from brain-derived protein levels in serum or CSF. High serum levels of neuron-specific enolase (NSE) in comatose patients at 24 and 48 hours after cardiopulmonary arrest predict poor neurologic outcome.180 Martens et al. found that serum concentrations of the astrocyte-derived protein S-100B were superior to serum NSE (as well as CSF S-100B and NSE) in predicting which adult patients would regain consciousness after cardiac arrest.181 A study used a multimodal approach, combining serum concentrations of NSE, the astrocyte-derived protein S-100B, and somatosensory-evoked potentials (SSEPs), prospectively in 27 adult patients after cardiac arrest and were able to predict all patients that did not regain consciousness.182 Serum NSE is increased in children after cardiac arrest and peaks at 24 hours.183 NSE and other biomarkers may be useful in determining responsiveness to therapeutic interventions (e.g., it has been reported that decreasing serum NSE occurs mainly in hypothermic [vs. normothermic] adults after cardiac arrest and is predictive of outcome).184 Other biomarkers showing promise in case series are procalitonin,185,186 alpha II-spectrin breakdown products,187 serum glial fibrillary acidic protein,188 and brain natriuretic peptide.189
Studies have used SSEP monitoring in an attempt to provide early prognostic information about outcome after cardiopulmonary resuscitation. Madl et al. tested median nerve SSEPs in 66 adults after successful resuscitation from cardiac arrest. They reported that the presence or absence and the latency of the cortical N70 peak reliably differentiated between bad and favorable outcome with 100% predictive ability.190 Testing was performed within 48 hours of resuscitation, which is a clinically relevant time frame. Auditory evoked response testing has been used in children with cardiac arrest after a submersion accident.191 Normal evoked responses were observed in all children who recovered neurologically intact. Children who recovered with significant handicaps demonstrated reduction in wave V amplitude over time, and prolonged wave I-V inter-peak latencies. In adults, bilaterally absent N20 waves at 24 and 48 hours have a reported specificity of 100% in predicting permanent coma after cardiac arrest.192
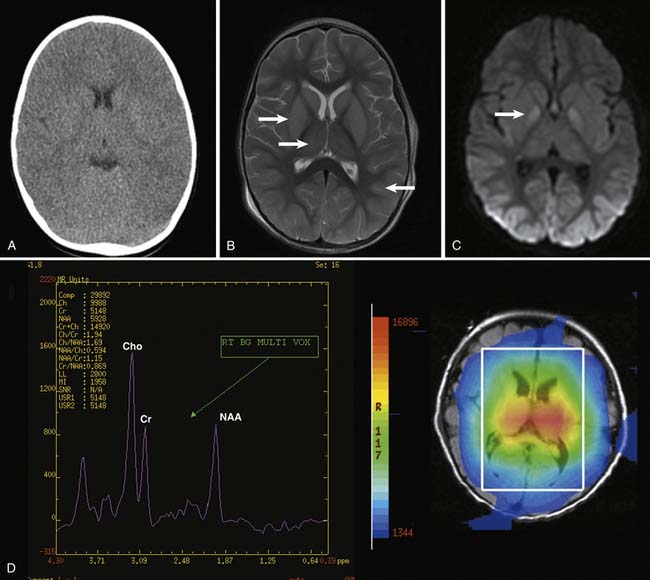
The utility of computed tomography (CT) of the head and other neuroimaging modalities after cardiac arrest is unknown (Figure 62-9). Head CT takes only a few minutes and is useful for evaluation of intracranial lesions contributing to the cause of arrest, but requires the critically ill subject to be transported out of the ICU and uses ionizing radiation. In a large retrospective case series of children with near-drowning, children with any abnormal CT findings (e.g., loss of gray-white differentiation, infarction) within the first 24 hours of admission died.193 Ninety-six percent of children with an initially normal CT on the first day that had a subsequent abnormal CT either died or remained in a coma. Using a novel scoring system assessing primarily the cortex and basal ganglia, a good correlation between early magnetic resonance imaging score and neurologic outcome was reported.194 Using magnetic resonance spectroscopy, decreases in the brain metabolite N-acetylaspartate and increases in lactate in the basal ganglia and cortex are can assist in outcome determination in children after near-drowning and cardiac arrest.147,195,196
< div class='tao-gold-member'>





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


