CHAPTER 28 Hypertensive Emergencies
Definition
HYPERTENSIVE EMERGENCIES are characterized by severe elevations in blood pressure (BP) (>180/120 mm Hg) complicated by evidence of impending or progressive target-organ dysfunction.1 The major types of hypertensive emergencies are summarized in Table 28-1. In reality, however, it is not the absolute level of BP that is hazardous, but the rate of rise of BP. Patients with chronic hypertension can tolerate much higher BP than previously normotensive persons. Diastolic BP (DBP) of 140 mm Hgor higher may be well tolerated in a patient with chronic hypertension without symptoms, whereas a DBP of 110 mm Hg may be devastating in a previously normotensive woman with preeclampsia or a child with acute glomerulonephritis. The diagnosis of hypertensive emergency, whatever the BP level, requires immediate reduction of BP, generally with an intravenous antihypertensive agent in an intensive care unit for continuous monitoring of BP. In contrast, a severe elevation in BP without evidence of progressive end-organ damage, considered an urgent hypertensive crisis, can usually be managed with oral medication in an outpatient setting. In this case, gradual reduction in BP over a period of 24 to 48 hours is usually sufficient.
Table 28–1 Types of Hypertensive Emergencies
Included among hypertensive emergencies is the syndrome of malignant or accelerated hypertension. As first described by Volhard and Fahr in 1914, malignant hypertension was characterized by severe hypertension, retinopathy with papilledema, renal insufficiency, fibrinoid necrosis of renal arterioles, and a rapidly progressive and fatal clinical course.2 Later, the definition was generalized to include severe hypertension accompanied by papilledema (grade IV Keith-Wagener retinopathy). In contrast, accelerated hypertension was considered severe elevation of BP in the presence of retinal hemorrhages and exudates but without papilledema (grade III Keith-Wagener retinopathy). Subsequent studies showed that severe hypertension complicated by retinal hemorrhages and exudates with or without papilledema had similar clinical features and prognosis, so that the terms malignant and accelerated hypertension are used interchangeably.3 Presently, these terms are not used, and, instead, hypertensive emergency is used to refer to the syndrome of elevated BP complicated by acute end-organ damage, including grade III or IV retinopathy.
Incidence and Prevalence
In spite of improvements in the diagnosis and treatment of hypertension, the control rate remains low. In the National Health and Nutrition Examination Survey III (NHANES III) only 37% of patients with hypertension had controlled BP levels.4 However, widespread outpatient antihypertensive treatment has reduced the incidence of hypertensive emergencies, and nonadherence to prescribed medication now accounts for most cases.5 Hypertensive emergencies occur in approximately 1% of hypertensive patients.6 In a Brazilian retrospective study of 76,723 patients admitted to the emergency department, 273 (0.35%) were considered hypertensive urgencies and only 179 (0.23%) had a diagnosis of hypertensive emergency.7 Similarly, 118 of 17,952 (0.65%) patients admitted to an emergency department in Spain were diagnosed as having a hypertensive crisis (defined as BP =210/120 mm Hg), but only 28 (0.15%) were diagnosed as a hypertensive emergency.8 In an Italian series, 108 hypertensive emergencies comprised 0.76% of visits to the internal medicine section of an emergency department during 1 year.9 The estimated prevalence of hypertensive crisis in the general hypertension population is 1% in the United States.10 Two major university hospitals in the United States have reported similar findings: in New York, 0.6% of 9851 medical service admissions were for hypertensive emergency, whereas in Miami, 1.1% of 2898 medical emergency department visits resulted in hospital admissions for hypertensive emergency.11,12 This contrasts with a 7% incidence of accelerated hypertension with papilledema in patients with untreated essential hypertension in the era before the development of effective antihypertensive therapy.13,14 The reduction in incidence of hypertensive emergencies has generally been attributed to the success of antihypertensive therapy in preventing progression from the early stages of hypertension to more advanced disease with target-organ damage. Poor access to health care and noncompliance to medications are generally associated with hypertensive crisis and should be assessed in patients having hypertensive emergencies before hospital discharge.
Etiology and Pathogenesis
The etiology of malignant or accelerated hypertension is unknown, but many conditions are related to hypertensive emergencies and urgencies (Table 28-2). The degree of BP elevation does not correlate closely with the severity of end-organ deterioration, and it is uncertain whether malignant or accelerated hypertension is a nonspecific consequence of very high BP or is triggered by a specific constellation of neurohumoral factors and cytokines.14–17
Table 28–2 Conditions Related to Hypertensive Emergencies
| Essential hypertension |
| Renal parenchymal disease |
| Renovascular disease |
| Pregnancy |
| Endocrine |
| Drugs |
| Autonomic hyperactivity |
| Central nervous system disorders |
From Vaughan CJ, Delanty N: Hypertensive emergencies. Lancet 2000;356:411-417.
The primary abnormality in patients with hypertensive emergencies is the impaired autoregulatory capacity (i.e., the ability of blood vessels to dilate or constrict to maintain normal perfusion, particularly in the cerebral and renal beds). An initial abrupt rise in vascular resistance in response to excess production of catecholamines, angiotensin II, vasopressin, aldosterone, thromboxane and/or endothelin, or deficient production of endogenous vasodilators—such as nitric oxide and prostacyclin—seems to precipitate increased vasoreactivity and resultant hypertensive emergency (Fig. 28-1).17
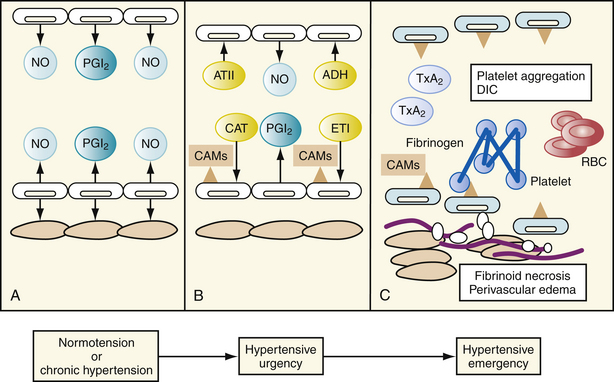
Autoregulatory function is compromised, resulting in end-organ ischemia, which triggers the release of additional vasoactive substances, thus initiating a vicious cycle of further vasoconstriction, myointimal proliferation, and end-organ ischemia.18 Preclinical studies have provided evidence that activation of renin-angiotensin-aldosterone system (RAAS) plays an important role in the pathophysiology of severe hypertension, leading to hypertensive crisis. Animals that are double transgenic for the human renin and angiotensinogen genes develop severe hypertension plus inflammatory vasculopathy similar to that seen in severe human hypertension.19 Angiotensin II has direct cytotoxic effects on the vessel wall through activation of expression of genes for proinflammatory cytokines and the transcription factor nuclear factor κB (NF-κB).20,21 The RAAS also induces expression of proinflamatory cytokines and vascular cell adhesion molecules, which may contribute to the vascular injury and target-organ damage.17 Arteriolar fibrinoid necrosis ensues, precipitating endothelial damage, platelet and fibrin deposition, and thromboxane release. Some studies have also reported a relation between genetic polymorphisms in components of the RAAS and hypertensive crisis. For example, the DD and ID genotypes of the angiotensin-converting enzyme (ACE) gene are more frequent in patients having malignant hypertension or hypertensive crisis.22,23
Malignant or accelerated hypertension is more prevalent among patients with secondary hypertension, particularly hypertension secondary to renal artery stenosis or renal parenchymal disease, than among essential hypertensive patients. However, essential hypertension is the most common underlying cause of malignant or accelerated hypertension because of its greater prevalence. Based on postmortem analysis, Kincaid-Smith found that of 124 cases of malignant hypertension, 44% occurred in the setting of primary hypertension; 13% had chronic glomerulonephritis; 9% had polyarteritis nodosa; and 6% had unilateral renal artery stenosis.24 In a study of 123 patients without evidence of primary renal parenchymal disease referred to a tertiary care center for evaluation of severe hypertension with grade III or IV retinopathy, Davis and associates found that 43% of the white patients and 7% of the black patients had renal artery stenosis documented by renal arteriography.25 Of the 242 patients with malignant hypertension identified by Lip and associates, 97 (40%) had secondary hypertension, most commonly related to renal parenchymal disease. Twenty-five patients (10%) had pregnancy-induced hypertension.26 Aldosterone excess has also been detected in patients with hypertensive crisis. Labinson and colleagues reported eight patients with a clinical diagnosis of primary aldosteronism whose course was complicated by hypertensive emergencies.27 All of these responded well to laparoscopic adrenalectomy or medical treatment with aldosterone receptor blockers. Accordingly, any patient who has experienced malignant or accelerated hypertension should undergo evaluation for secondary hypertension.
Diagnosis
The common presenting symptoms and target organ complications of hypertensive emergencies are listed in Table 28-3.26 The physician evaluating a patient for hypertensive emergency should focus on the history, physical examination, and laboratory evaluation to assess the severity of BP elevation and related end-organ damage.16 This includes a determination of the duration, severity, and level of control of preexisting hypertension, antihypertensive drug treatment, and the extent of preexisting end-organ damage. The presence of concomitant illnesses and all medications, including prescription drugs, over-the-counter preparations such as sympathomimetic agents, and illicit drugs such as cocaine, should be noted. The physician should assess specific symptoms suggesting end-organ damage such as chest pain (myocardial ischemia or aortic dissection), back pain (aortic dissection), dyspnea (pulmonary edema or congestive heart failure), and neurologic symptoms (hypertensive encephalopathy).
Table 28–3 Most Common Presenting Symptoms and Complications of Hypertensive Emergencies
| Symptoms/Complications | Frequency |
|---|---|
| 26% | |
| 12% | |
| 10% | |
| 8% | |
| 7% | |
| 5% | |
| 10% | |
| 11% | |
| 10% | |
| 4% | |
| 4% | |
| 32% | |
| 61% |
From Lip GYH, Beevers M, Beevers G: The failure of malignant hypertension to decline: a survey of 24 years’ experience in a multiracial population in England. J Hypertens 1994;12:1297-1305.
The signs of hypertensive emergency that should be assessed on physical examination are listed in Table 28-4. Supine and upright BP should be measured to assess volume status. The BP should also be measured in both arms because a significant difference increases the suspicion of aortic dissection. The ocular fundi should be examined for evidence of acute hypertensive retinopathy. The heart and lungs should be auscultated for signs of acute left ventricular dysfunction. The presence of an abdominal bruit suggests renal artery stenosis and palpation of the abdomen may reveal evidence of an aortic aneurysm. Discrepancies in peripheral pulses and the murmur of aortic regurgitation suggest aortic dissection. The neurologic examination may yield evidence of encephalopathy or stroke, including disorientation, localized weakness, and visual field deficits.
Table 28–4 Physical Examination in Patients with Hypertensive Emergency
| Blood pressure measurements |
| Fundoscopy |
| Heart and lungs auscultation |
| Abdomen |
| Palpation of peripheral pulses |
| Neurologic examination |
Basic diagnostic evaluation for patients with hypertensive crisis is listed in Table 28-5. Urinalysis and determination of serum electrolytes, urea nitrogen, and creatinine levels should be obtained to confirm or exclude acute renal insufficiency; a complete blood cell count with peripheral blood smear should be performed to look for evidence of microangiopathic anemia; and a serum glucose test should be done to identify concomitant diabetes.5,17 An electrocardiogram to help identify or rule out myocardial ischemia and chest radiograph to look for pulmonary edema secondary to left ventricular failure and for widening of the mediastinum secondary to thoracic aortic dissection are indicated.5,17 In some cases, measurement of plasma renin activity and aldosterone drawn at the time of admission can be useful in making a retrospective diagnosis. If aortic dissection is suspected, computed tomography, magnetic resonance imaging, and transesophageal echocardiography may be needed.28
Table 28–5 Diagnostic Tests for Evaluation of Patients with Hypertensive Crisis
| Blood |
| Urine analysis |
| Electrocardiogram |
| Chest radiography |
Any evidence of acute or rapidly progressive end-organ deterioration in the history, physical examination, or laboratory evaluation distinguishes a hypertensive emergency from an urgent hypertensive crisis.6 As soon as the physician diagnoses or suspects target organ damage from the history, physical examination, or laboratory analysis, the patient should be placed on a cardiac monitor and have intravenous access established. BP reduction (usually with an intravenous drug) should begin during the arrangements for hospital admission.
Treatment
Patients with a hypertensive emergency should be admitted to an intensive-care unit and require immediate but controlled reduction in BP to protect end-organs from further damage. In general, the goal of therapy is to lower the mean arterial pressure by approximately 25% or to reduce the DBP to 100 to 110 mm Hg over a period of several minutes to hours, depending on the clinical situation.1,6 Precipitous reductions in BP and rapid reductions to normotensive or hypotensive levels should be avoided because they may provoke end-organ ischemia or infarction. BP should be maintained at the initial target level for 2 to 6 hours. If the BP is tolerated and the patient is clinically stable, further gradual reductions can be implemented in the next 24 to 48 hours.1 Ischemic stroke and aortic dissection are exceptions. There is no evidence from clinical trials to support immediate antihypertensive treatment in patients with ischemic stroke.29 Oral or intravenous antihypertensive treatment should be instituted if DBP greater than 120 mm Hg or if the patient is eligible for thrombolysis.30 Aortic dissection is a dramatic and fatal complication, and these patients should have their systolic BP (SBP) reduced to less than 100 mm Hg if tolerated.1 Therapy for acute dissection aims to reduce stress on the aortic wall by lowering BP and heart rate. These goals are achieved with a combination of β-blockers (labetalol is most commonly used) and vasodilators. Nitroprusside can cause reflex tachycardia and thus β-blockers should be started first.17
The goal of therapy is to interrupt the cycle of impaired autoregulatory capacity and vasoreactivity by reducing peripheral vascular resistance.6 Sodium nitroprusside is the drug of choice for the treatment of most hypertensive emergencies because it allows for the controlled reduction of BP. In all situations in which an antihypertensive agent is being delivered intravenously, constant BP monitoring, preferably through an arterial line, is mandatory. Most patients who have a hypertensive crisis are volume-depleted, presumably secondary to a pressure-related diuresis. Accordingly, diuretics should be reserved for patients in whom there appears to be fluid overload, such as pulmonary edema.
Parenteral antihypertensive agents should be gradually withdrawn after BP has been controlled for 12 to 24 hours, with concomitant introduction of oral agents. Evaluation for secondary hypertension may be started during hospitalization while oral antihypertensives agents are titrated. Attempting to ensure adherence to antihypertensive therapy during long-term follow-up is an important step to prevent recurrent hypertensive crisis.5
Prognosis
The development of successful antihypertensive therapy has greatly improved the prognosis of patients with hypertensive emergencies.5 In 1939, Keith and colleagues found that mean survival of patients with hypertension and grade IV retinopathy was 10.5 months, with no survivors at 5 years.31 More recently, authors have reported that the survival of patients with malignant hypertension approaches that of patients with uncomplicated primary hypertension.32,33 Using life-table analysis, the estimated survival of patients with malignant hypertension was 18 years, versus 21 years in patients with uncomplicated hypertension.32 The leading cause of death (50%) in patients with accelerated hypertension was myocardial infarction. In Birmingham, UK, 74% of 315 patients with malignant hypertension were alive after 5-year follow-up and renal failure was the most common cause of death.33 Acute target-organ damage secondary to hypertensive emergency (e.g., acute renal failure may return toward normal if BP is controlled in the long term).5 An exception to this rule is eye and central nervous system damage, which tends to be irreversible.
Specific Hypertensive Emergencies
Hypertensive Encephalopathy
Hypertensive encephalopathy is the most difficult neurologic emergency to diagnose and frequently it is a diagnosis of exclusion.17 With better diagnosis and treatment of primary hypertension, hypertensive encephalopathy is infrequent. However, prompt recognition and treatment is essential to prevent progression to hemorrhage. This becomes a particularly difficult issue when hypertensive encephalopathy occurs in previously normotensive persons, as with preeclampsia or “crack” cocaine abuse. Hypertensive encephalopathy is also associated with secondary causes of severe hypertension, such as renal disease, immunosuppressive therapy,34,35 erythropoietin use,36 and thrombotic thrombocytopenic purpura.37
Ordinarily cerebral blood flow is autoregulated (i.e., cerebral resistance vessels constrict and dilate in response to changes in systemic pressure) to maintain a constant perfusion pressure (Fig. 28-2).17,38–41 In normotensive subjects, perfusion pressure is maintained over a wide range of mean arterial pressure (60 to 120 mm Hg). With chronic hypertension, the mean BP range over which cerebral perfusion is maintained is shifted upward (160 to 180 mm Hg), such that persons with long-standing hypertension can tolerate greater elevations in BP without compromising cerebral perfusion. When elevations in systemic BP exceed the brain’s ability to locally autoregulate cerebral perfusion, overperfusion ensues, resulting in cerebral edema, small vessel damage, and microinfarctions. As indicated earlier, breakdown in autoregulation of cerebral perfusion occurs at much lower BP levels in previously normotensive subjects than in persons with chronic hypertension.
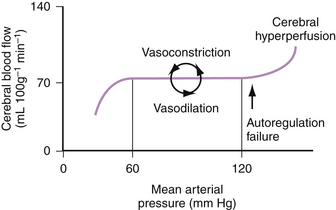
Figure 28-2 Autoregulation of cerebral blood flow.
(From Vaughan CJ, Delanty N: Hypertensive emergencies. Lancet 2000;356:411-417.)
Hypertensive encephalopathy most commonly develops over a period of several days. The patient usually complains of headache, nausea, visual changes, or focal neurologic symptoms. Physical examination generally reveals disorientation, obtundation, focal neurologic signs, seizures, and retinopathy, including papilledema. Hypertensive encephalopathy is a diagnosis of exclusion after ruling out stroke, subarachnoid hemorrhage, seizure disorder, encephalitis, and drug overdose.17
The drug of choice for treatment of hypertensive encephalopathy is nitroprusside (Table 28-6). It should be titrated to reduce mean arterial pressure by about 20% or to a DBP of 100 mm Hg within the first hour.17 Particular caution is needed in elderly patients and in those with preexisting hypertension in whom overagressive reduction in BP may be accompanied by worsening of neurologic status and even stroke. Neurologic function should improve with BP reduction. If not, a diagnosis other than hypertensive encephalopathy should be entertained. Labetalol can be used as an alternative to nitroprusside. Agents that suppress central nervous system function, such as β-blockers and central nervous system active agents, and agents that increase cerebral blood flow, such as nicardipine and nitroglycerin, should be avoided.
Table 28–6 Types of Hypertensive Emergency and Treatment Recommendations
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes: Mechanism, Significance, and Prognosis
 Hemodynamically Unstable Presentations of Congenital Heart Disease in Adults
Hemodynamically Unstable Presentations of Congenital Heart Disease in Adults
 Conduction Disturbances in Acute Myocardial Infarction
Conduction Disturbances in Acute Myocardial Infarction
 Antiplatelet Therapy
Antiplatelet Therapy
 Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Pacemaker and Implantable Cardioverter-Defibrillator Emergencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


