101 Hepatic Encephalopathy
Hepatic encephalopathy encompasses a spectrum of neuropsychiatric abnormalities that occur in patients with liver disease in the absence of other brain disease.1–2 The spectrum includes personality changes, impaired mental function, motor abnormalities (asterixis, tremor, hyperventilation, hyperactive reflexes), and altered consciousness. A consensus panel of experts proposed classification of hepatic encephalopathy into type A, associated with acute liver failure; type B, associated with portal-systemic bypass without intrinsic liver disease; and type C, associated with chronic liver disease.3
The encephalopathy accompanying acute hepatic failure (type A) is commonly associated with cerebral edema and increased intracranial pressure (ICP), exhibits abrupt onset with a short prodrome and rapid progression, and often ends with the death of the patient.4–6 Patients sequentially experience drowsiness, delirium, agitation or convulsions, decerebrate rigidity, unresponsiveness, and deep coma within a comparatively short period of time, usually hours to days. Irreversible neurologic damage may occur as a result of brain ischemia or herniation. Patients who develop coma in the setting of acute liver failure have a grave prognosis; fewer than 20% survive without hepatic transplantation.7
In patients with chronic liver disease, encephalopathy (type C) develops insidiously and often is heralded by a change in mental or behavioral status. Encephalopathy may be episodic, persistent, or minimal and subclinical.3 Episodes are sporadic, characterized by exacerbations and remissions, and generally are precipitated by inciting events.1–2 Although the initial manifestation of portosystemic encephalopathy (PSE) is usually a subtle change in mentation, neurologic dysfunction may progress and be classified according to confusion, lethargy, and even coma (Table 101-1).8 Neurologic signs vary and fluctuate but usually include asterixis, hyperreflexia, clonus, and an extensor plantar response. Causes of PSE may not always be apparent, but azotemia, sepsis, gastrointestinal (GI) bleeding, dehydration, electrolyte imbalances, and sedatives are frequent precipitants (Box 101-1). In some patients, chronic encephalopathy may not be clinically obvious, but only detectable by psychometric testing. By these tests, about two-thirds of cirrhotic patients with portal hypertension have unsuspected subclinical hepatic encephalopathy.9–12 Patients who undergo portal-systemic shunt or bypass, either surgical or transjugular intrahepatic portosystemic shunt (TIPS), often develop encephalopathy (type B) which is similar to the encephalopathy experienced by patients with chronic liver disease.
TABLE 101-1 Stages of Encephalopathy in Chronic Liver Disease
| Stage | Clinical Signs |
|---|---|
| Stage I | Mental slowness, euphoria or anxiety, shortened attention span, impaired calculating ability |
| Stage II | Lethargy or apathy, inappropriate behavior, personality change, more obvious problems with calculations |
| Stage III | Lethargic, somnolent, marked confusion and disorientation, but responds to verbal stimuli |
| Stage IV | Coma, patient may or may not respond to noxious stimuli |
Patients with chronic liver disease rarely, if ever, demonstrate cerebral edema, regardless of the stage of encephalopathy.
 General Principals
General Principals
No single abnormality of hepatic or neurologic metabolism adequately explains all of the clinical, biochemical, physiologic, or experimental findings of encephalopathy occurring in patients or animal models.1–26 Abnormalities of multiple neurotransmitters including glutamate, γ-aminobutyric acid (GABA), dopamine, serotonin, and opioids have been described, and plasma levels of a wide array of potential neurotoxins (ammonia [NH3], GABA, short-chain fatty acids, methanethiols) are increased (Box 101-2).13–14 Despite this seeming confusion, several lines of investigation focus on NH3 as a key factor in the pathogenesis of hepatic encephalopathy. Ammonia accumulation deranges glutamate and glutamine metabolism in the central nervous system (CNS) and alters the metabolism of GABA and its function as an inhibitory neurotransmitter. In addition, benzodiazepine receptors in the CNS are physically linked to GABA receptors. The latter finding provides an explanation for the increased sensitivity of patients with liver disease to the sedative and hypnotic effects of benzodiazepines and a rationale for use of benzodiazepine antagonists in the treatment of PSE. Hepatic encephalopathy occurring in the setting of either acute liver failure or chronic liver disease is also associated with marked changes in CNS glial cells on neuropathologic examination. Encephalopathy of acute liver failure is characterized by astrocytic swelling, but chronic encephalopathy is characterized by Alzheimer type II astrocytosis.15
Cerebral Blood Flow
In acute liver failure, the brain is potentially subject to hypoxic injury due to complications such as systemic arterial hypotension, respiratory failure, and reduction in cerebral blood flow that accompanies cerebral edema and intracranial hypertension. Therapy is often directed at optimal oxygenation, maintenance of cerebral perfusion pressure (goal > 40 mm Hg), and reduction of ICP (goal < 20 mm Hg).4,9 Paradoxically, increases in cerebral blood flow, however, may aggravate cerebral edema and worsen neurologic damage. In humans with acute liver failure, cerebral blood flow has been measured primarily by the xenon-133 washout technique.16–20 These data suggest that cerebral blood flow is initially relatively low but then increases with increasing blood concentration of NH3, which decreases cerebral arteriolar tone.
Cerebral Glucose and Oxygen Metabolism
Brain energy metabolism is unique in that glucose is the only substrate under normal physiologic conditions, and its uptake and utilization by the brain is independent of insulin.21–25 Under stress, the brain can utilize β-hydroxybutyrate and acetoacetate. Ammonia accumulation during hepatic failure in humans or in experimental models of hyperammonemia is associated with altered cerebral glucose metabolism. In early acute liver failure, prior to onset of intracranial hypertension, cerebral glucose metabolism and cerebral oxygen consumption are proportionately diminished.21 There is no evidence of cerebral hypoxia, implying that the reduced glucose and oxygen utilization reflect diminished metabolic demand by the brain at this early stage. Cerebral lactate uptake is increased despite sufficient glucose delivery and preserved oxidative metabolism. Acute short-term mechanical ventilation, resulting in moderate reduction in PCO2 and cerebral blood flow, does not adversely affect oxidative brain metabolism. Thus, prior to the development of intracranial hypertension, cerebral glucose and oxygen metabolism are reduced, but these changes are consistent with normal aerobic metabolism and physiologic regulation. After development of intracranial hypertension, oxygen metabolism remains reduced, but measurements of cerebral glucose utilization vary from reduced rates to increased rates, and glycolysis may be accelerated.22–2325 These findings suggest that progression of acute liver failure and development of intracranial hypertension are associated with relative cerebral hypoxia and a switch to anaerobic metabolism.
Ammonia Hypothesis
The ammonia hypothesis states that the major mechanism of hepatic encephalopathy is excessive accumulation of NH3, which induces neuronal metabolic derangements but also promotes astrocytic swelling.26 In addition, NH3 perturbs cerebral nitric oxide metabolism, which can mediate some of the effects.27 Studies using positron-emission tomography (PET) and magnetic resonance spectroscopy have demonstrated an increase in cerebral metabolic rate and increased permeability of the blood-brain barrier for NH3.28–34 Blood NH3 originates mainly from four sources: intrahepatic deamination of amino acids, extrahepatic metabolism of nucleotides, gut metabolism of glutamine, and bacterial degradation of intestinal protein and urea.35 More than 50% of blood NH3 is derived from the latter source. NH3 is normally metabolized by the liver to either urea or glutamine by the actions of carbamoyl-phosphate synthetase I (the initiating enzyme of the urea cycle) and glutamine synthetase, respectively. Patients with hepatic failure have impaired NH3 metabolism related to a reduction in liver metabolism and an increase in portal-systemic shunting. As a result, an elevation in blood NH3 concentration is a characteristic feature of severely impaired hepatic function.
Certain clinical and experimental observations link the increase in blood NH3 concentration to hepatic encephalopathy.13,15,36–37 Hyperammonemia and elevated concentrations of NH3 within the cerebrospinal fluid (CSF) are features of acute and chronic hepatic encephalopathy, Reye syndrome, deficiencies of urea cycle enzymes, and sodium valproate toxicity. In cirrhotics or patients with portocaval shunts, ingestion of NH3-generating substances (proteins, amino acids, urea, ammonium salts) may precipitate encephalopathy. In animal models, chronic administration of ammonium salts results in Alzheimer type II astrocytosis, a change indistinguishable from that observed in patients with chronic hepatic encephalopathy.15
Glutamine-Glutamatergic Neurotransmitter System
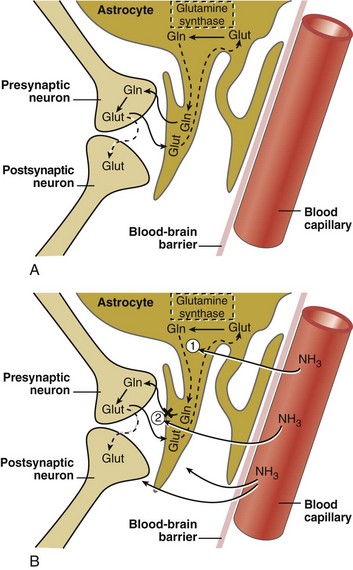
The glutamatergic excitatory neurotransmitter system in the CNS is markedly altered in patients with both acute and chronic liver disease and in animal models of hepatic encephalopathy.6,26–27 CNS astrocytes are a major regulatory cell in the glutamatergic system.15 Normally the astrocyte avidly takes up excess glutamate from the synaptic cleft (against a 3000-10,000 fold concentration gradient), an important function that terminates glutamate-induced neuroexcitation. Once glutamate is taken up by the astrocyte, it is metabolized to glutamine via the action of glutamine synthetase, which utilizes blood-derived NH3 (Figure 101-1). The hyperammonemia of liver failure favors the formation of glutamine but also impairs the release of glutamine from the astrocyte. The accumulation of osmotically active glutamine in the astrocyte is associated with cell swelling. Normally, glutamine is actively extruded from the astrocyte and then taken up by presynaptic nerve terminals for conversion back to glutamate and subsequent utilization in neurotransmission. Under the conditions of liver failure and hyperammonemia, glutamate uptake into neurons and astrocytes is diminished, and glutamate accumulates in the extracellular fluid. Clinically, levels of glutamine and glutamate increase in CSF fluid during hyperammonemic states, and CSF concentrations of glutamine correlate loosely with the stage of encephalopathy. In animal models of acute encephalopathy, blockade of glutamine production by an inhibitor of glutamine synthetase, methionine sulfoximine, decreases cerebral edema and reduces astrocyte swelling.14 Production of NH3 from the intestine is reduced by orally administered nonabsorbable antibiotics such as rifaximin and neomycin, as well as nonabsorbable disaccharides including lactulose, lactitol, and lactose (in lactase-deficient patients). These treatments lower plasma NH3 concentration and improve subjective and objective measures of encephalopathy.
Other clinical and experimental observations refute the link between NH3 and hepatic encephalopathy. Blood levels of NH3 are elevated in cirrhotic patients regardless of the presence or absence of encephalopathy. Some patients with hepatic encephalopathy have normal blood levels of NH3. The grade of hepatic encephalopathy does not correlate with the blood concentration of NH3. Seizures and hyperexcitability are commonly observed in animal models of NH3 intoxication and in human congenital hyperammonemia but are rarely observed in patients with chronic hepatic encephalopathy. Administration of ammonium chloride to cirrhotic patients induces a mild hyperkinesis but fails to exacerbate typical chronic encephalopathy.13
γ-Aminobutyric Acid-Benzodiazepine Receptor Hypothesis
GABA is an inhibitory neurotransmitter found throughout the CNS.38 The GABA hypothesis states that an excess of GABA or increased sensitivity to GABA is responsible for hepatic encephalopathy.14,38–39 Observations in rabbits with galactosamine-induced hepatic failure provided the initial support for this hypothesis. GABA originates from the intestine, and plasma levels increase in hepatic failure due to inadequate hepatic extraction. During acute liver failure, the blood-brain barrier becomes more permeable, and increased amounts of GABA enter the CNS. Once in the brain, GABA binds to its receptor to produce neuroinhibition and clinical encephalopathy. A key component to understanding the relationship of GABA and benzodiazepines was the recognition that the GABA receptor was tightly linked and modulated by the benzodiazepine receptor.40–43 Binding of benzodiazepines to the benzodiazepine receptor induces a conformational change in the GABA receptor enhancing the binding of GABA and neuroinhibition. Activation of the GABA receptor opens a chloride channel, the third component of the GABA receptor complex (Figure 101-2). In summary, GABA-induced inhibition of neurotransmission is enhanced by binding of benzodiazepines or related compounds to the benzodiazepine receptor, increasing the number of GABA receptors, increasing the activity of the GABA receptor, or opening the GABA-associated chloride channel.
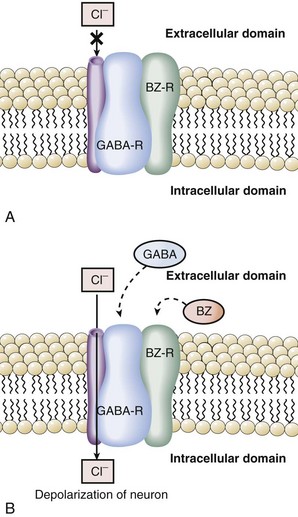
The GABA hypothesis predicts that benzodiazepines would increase the severity of hepatic encephalopathy, and benzodiazepine antagonists such as flumazenil might ameliorate hepatic encephalopathy. Clinical experience clearly suggests that cirrhotic patients, especially those with encephalopathy, are particularly sensitive to the amnesic and sedative effects of benzodiazepines. In our experience, use of benzodiazepines and other sedative/hypnotics is a common reason for exacerbations of hepatic encephalopathy. Recent studies have demonstrated that patients with hepatic encephalopathy have increased plasma levels of benzodiazepines or “natural” benzodiazepine-like compounds that then may act as “false neurotransmitters.”44–47 Some have suggested that patients with hepatic encephalopathy are particularly sensitive to GABA neuroinhibition, owing to an increase in background benzodiazepine stimulation of the GABA receptor.
Taurine
Taurine is an inhibitory neurotransmitter which is increased in brains of animal models of experimental hepatic encephalopathy and in the CSF of primates with encephalopathy secondary to portocaval shunts. Plasma levels of taurine are greatest in patients with the greatest degrees of encephalopathy, suggesting that this inhibitory neurotransmitter may be involved in hepatic encephalopathy.48 Additional neurotransmitters that may be altered in hepatic encephalopathy include endogenous opioids and melatonin.
Methanethiols
Interest in methanethiol as a potential neurotoxin began with the finding of methanethiol in the urine of a patient with fetor hepaticus. Subsequently, levels of methanethiol, 4-methylthio-2-oxobutyrate, and methanethiol-mixed disulfides were found to be elevated in the plasma of cirrhotic patients.49 It was suggested that these compounds may exacerbate the toxic effects of NH3 and short-chain fatty acids. However, blood levels of methanethiols are similar in deeply comatose patients and those with only mild cerebral dysfunction, and there is little correlation between grade of encephalopathy blood levels of methanethiol.
Manganese, Zinc
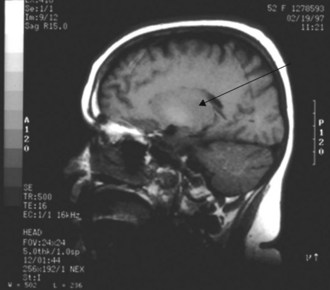
The liver is responsible for manganese excretion, and liver disease is associated with manganese accumulation. Magnetic resonance imaging (MRI) studies of the brain in patients with cirrhosis reveal pallidal hyperintensity on T1-weighted images (Figure 101-3), which correlates with the presence of extrapyramidal signs and symptoms and blood levels of manganese.50 Manganese concentrations in the globus pallidus are markedly increased in autopsy studies of cirrhotics who died in hepatic coma. PET imaging reveals reduced cerebral glucose utilization in these areas. Such findings suggest a relationship between manganese, brain hypometabolism, and some of the neuropsychiatric and motor abnormalities of hepatic encephalopathy.
Zinc deficiency is common in long-standing cirrhosis. Its importance to the pathogenesis of hepatic encephalopathy is unknown, and three randomized controlled trials of zinc supplementation have yielded conflicting results as regards improvement in encephalopathy (one positive, two negative).51–53
 Encephalopathy in the Setting of Acute Hepatic Failure
Encephalopathy in the Setting of Acute Hepatic Failure
Definition
Acute liver failure is defined by the development of coagulopathy (prothrombin time [PT] >20 sec prolonged with an international normalized ratio [INR] >1.5) in a patient with acute hepatitis who lacks underlying chronic liver disease (exception in Wilson’s disease).54–56 Patients with acute liver failure usually have extreme elevations of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) with the initial injury (1000 to 5000 IU/L), often are jaundiced, and exhibit constitutional symptoms. They are at risk for encephalopathy, although most recover uneventfully. Progressive hepatic encephalopathy is a poor prognostic sign and signals the need for hepatic transplantation.
Etiology
There are approximately 2000 cases of acute liver failure in the United States each year.57,58 Among the most common causes are acetaminophen toxicity, other types of acute drug toxicity, and hepatitis A and B virus (HAV, HBV) infection. However, the second leading diagnostic category for fulminant hepatic failure (FHF) is cryptogenic, cause unknown (Table 101-2). Recent data obtained since 1998 indicate that over 50% of cases of FHF in the United States are due to acetaminophen overdosage (38%) or idiosyncratic drug reactions (~14%).58 Many cases of acetaminophen-induced liver failure are due to “therapeutic misadventure” due to self-administration of as little as 4 grams/day over several days in the setting of fasting and alcohol use. Sporadic cases of FHF due to both cocaine and Ecstasy have recently been described. FHF from mushroom poisoning occasionally occurs with inexperienced amateur mushroom fanciers. Infiltration of the liver with rapid progression of tumor growth can lead to FHF and has been described for cases of metastatic breast carcinoma, lymphoma, and melanoma. Biopsy of the liver is required to establish the latter diagnoses. FHF also may occur in the third trimester of pregnancy, related to acute fatty liver of pregnancy, HELLP syndrome, or disseminated herpes infection.
TABLE 101-2 Causes of Acute Liver Failure
| Acetaminophen | 20% |
| Cryptogenic | 15% |
| Non-acetaminophen drug toxicity | 12% |
| Hepatitis B | 10% |
| Hepatitis A | 7% |
| Autoimmune hepatitis | 6% |
| Wilson’s disease | 6% |
| Miscellaneous* | 24% |
* Budd-Chiari syndrome, herpes simplex, paramyxovirus, Epstein-Barr virus, amanita poisoning, ischemia, malignant infiltration.
Adapted from Schiodt FV, Atillasoy E, Shakil O et al. Etiologic factors and outcome for 295 patients with acute liver failure in the United States. Liver Transplant Surg 1999;5:29-34.
Prognosis
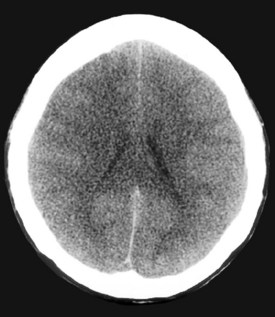
A major determinant of prognosis is the level of encephalopathy (see Table 101-1). Patients with acute liver failure who have progressed to higher stages of encephalopathy (stage III or IV) have the worst prognosis. Glasgow Coma Scale is useful in assessing need for transplantation.59 Cerebral edema on computed tomography (CT) scan of the brain is a late feature of progressive encephalopathy (Figure 101-4). Additional clinical features that indicate a poor prognosis include metabolic acidosis, renal failure, severe jaundice, or markedly prolonged prothrombin time.56,59–60 The likelihood of survival varies with the cause of acute liver injury. Patients with acetaminophen overdose have a relatively favorable outcome, and over 50% survive.61 Patients with fulminant HAV and HBV infection have an intermediate prognosis, and 30% to 50%62 survive. In contrast, patients with a fulminant presentation of Wilson’s disease or severe sporadic non-A, non-B, non-C hepatitis have a survival of less than 10%.63
General Clinical Management
All patients should be placed on needle (HBV, NANBV) and stool (HAV) precautions but not in isolation. Gloves should be worn when handling biological specimens, and specimens should be clearly labeled (Hepatitis Patient). All used instruments should be autoclaved or appropriately disposed. FHF due to acute viral hepatitis is not reversible by antiviral therapy, although some advocate use of lamivudine or related drugs for severe acute HBV. Corticosteroids are contraindicated, as they may increase the risk of developing chronic hepatitis and are ineffective in treatment of encephalopathy or cerebral edema in this setting. Removal of the offending drug, toxin, or alcohol is the mainstay of therapy of drug-induced and alcoholic hepatitis, respectively. N-acetylcysteine is an effective primary intervention for hepatic injury related to acetaminophen64 and is currently under investigation in the treatment of FHF due to other etiologies (Box 101-3).
Box 101-3
Use of N-Acetylcysteine in Treatment of Acetaminophen Overdose
Oral Dosing Schedule
Intravenous Dosing Schedule (Limited Availability in Research Centers)
Infection is a leading comorbidity in patients with acute liver failure.56–60 Blood, urine, and sputum should be cultured frequently (even in absence of fever or other signs of infection) and antibiotic therapy instituted only for positive cultures and directed against specific organisms. Fever is not a feature of most forms of acute liver injury and if present usually signifies concurrent infection. Febrile patients should be cultured immediately and treated empirically with antibiotics. The most common sources of infection are the respiratory tract, the urinary tract, and line sepsis. Currently, we use vancomycin with a broad-spectrum cephalosporin such as cefotaxime or a fluoroquinolone such as levofloxacin as initial treatment, and then tailor antibiotic use once results of cultures are known.
 Use of Intracranial Pressure Monitoring
Use of Intracranial Pressure Monitoring
Advantages
The use of ICP monitoring for managing high-grade encephalopathy in acute liver failure remains controversial. Several studies advocate ICP monitoring for its ability to provide important prognostic information about neurologic recovery after hepatic transplantation and in managing intracranial hypertension (ICH) while awaiting liver transplantation.65–67 Therefore, ICP monitoring could be considered for patients with high-grade encephalopathy (stage III or IV) who are waiting on the transplant list. Also, centers could consider ICP monitoring in nontransplant candidates if there is a reasonable likelihood of spontaneous recovery, such as in acetaminophen-induced acute liver failure. In addition to helping with the management of ICH, ICP monitoring also assists with close monitoring during the crucial perioperative period of liver transplantation, since it has been found that there is a transient increase in ICP that can last for about 12 hours postoperatively, after the dissection of native liver and the reperfusion of the graft.68
Disadvantages
However, the use of ICP monitoring may lead to severe complications such as intracranial hemorrhage in these already critically ill patients. In addition, several nonrandomized studies have failed to demonstrate an improvement in survival with the use of ICP monitoring.65–67 One prospective study of 92 out of 332 patients with acute liver failure, high-grade encephalopathy, and ICP monitoring found a 10.3% rate of intracranial hemorrhage.67 However, nearly half of the cases of intracranial bleeding were incidental radiographic findings without clinical consequence. Regardless of ICP use, 30-day survival post transplant was approximately 85%. Other reports confirm that bleeding complications from the placement of ICP monitoring devices in patients with acute liver failure are mostly mild and without clinical significance.65–6769 Use of ICP monitoring remains controversial, but experts agree that these devices should not be used in patients with mild hepatic encephalopathy (grades I or II) or in patients with evidence of cerebral herniation, hypotension, or imminent death.
 Management of Encephalopathy and Intracranial Hypertension
Management of Encephalopathy and Intracranial Hypertension
Encephalopathy is a hallmark of acute liver failure. It is also observed in patients with underlying chronic liver disease who sustain superimposed acute liver injury. The encephalopathy of acute hepatic failure is related to both metabolic factors, such as progressive elevation in blood NH3 concentration, and cerebral edema. Progressively worsening encephalopathy is an ominous clinical feature; development of grade III or IV encephalopathy may herald the death of the patient due to cerebral edema, increased ICP, and central herniation of the brain. Efforts to control the encephalopathy of acute liver failure are directed at preventing or resolving cerebral edema (Box 101-4).16,67,70–73 Because emerging evidence suggests that NH3 may play a role in the development of cerebral edema, we recommend that administration of protein should be limited to less than 40 grams/d in adults, and lactulose (20-40 g/d in divided doses) should be administered enterally to purge the bowel. However, one must exercise caution when using lactulose in the setting of FHF; dosing should be monitored carefully and adjusted to avoid excessive diarrhea and alterations in electrolytes and volume depletion. If oral (PO) lactulose is given simultaneously with intravenous (IV) mannitol, marked deficits of free water can develop, inducing severe hypernatremia. Although one recent study suggested that infusion of hypertonic (3%) saline to maintain serum sodium concentration between 145 and 155 mEq/L is beneficial,74 rapid shifts in sodium concentration have been associated with central pontine myelinolysis. Further discussion of hypertonic saline is provided later in this chapter. Administration of terlipressin or vasopressin may worsen intracranial hypertension and should be avoided.75
Box 101-4
Measures Used to Monitor and Control Cerebral Edema Due to Fulminant Hepatic Failure


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


