47 Heart-Lung Interactions
Ventilation can profoundly alter cardiovascular function. The boundaries of the cardiovascular unit’s responsiveness are defined by both cardiovascular and pulmonary factors. These limitations include myocardial reserve, circulating blood volume, blood flow distribution, autonomic tone, endocrinological responses, lung volume, intrathoracic pressure (ITP) and surrounding pressures for the remainder of the circulation. That positive-pressure ventilation may influence cardiovascular function in ways not seen during spontaneous ventilation was appreciated when positive-pressure ventilation was first introduced over 50 years ago1 and still results in new perspectives today.2
 Airway Pressure, Intrathoracic Pressure, and Lung Volume Relationships
Airway Pressure, Intrathoracic Pressure, and Lung Volume Relationships
Since positive-pressure ventilation was introduced, the concept of relating hemodynamic consequences to airway pressure was widely accepted.3,4 This oversimplification has been the source of much of the confusion in the clinical literature. A major source of such confusion rests in equating changes in airway pressure (Paw) with changes in both pleural pressure (Ppl) and lung volume. Physicians often equate Paw with the hemodynamic effects seen because (1) Paw can be measured easily at the bedside in patients receiving mechanical ventilation, (2) mean Paw reflects mean alveolar pressure, and (3) increases in Paw qualitatively reflect increases in both lung volume and Ppl. However, the association between Paw and other hemodynamically relevant factors (1) is highly variable as ventilatory patterns, airway resistance, and lung compliance change, (2) does not accurately reflect changes in pericardial pressure (Ppc), which is a primary determinant of transmural left ventricular (LV) pressure, and (3) may mislead the caregiver at the bedside into altering therapy based on these wrong assumptions. Numerous studies have demonstrated that the primary determinants of hemodynamic responses to ventilation are due to changes in intrathoracic pressure and lung volume, and not Paw.5 Thus, prior to examining heart-lung interactions, we shall address the relation between Paw, Ppl, Ppc, and lung volume. To simplify the discussion, we shall use the term intrathoracic pressure (ITP) to refer to nonspecific intrathoracic surface pressure. When specific intrapleural surface pressures are identified, they will be referred to either as the lateral chest wall, diaphragm, and juxtacardiac pleural pressures or pericardial pressure where appropriate.
Airway Pressure, Lung Volume, and Regional Pleural Pressures
During positive-pressure inspiration, increases in Paw parallel increases in lung volume. In the sedated and paralyzed patient at end-inspiration, only lung and thoracic compliance determine the relationship between Paw and lung volume. However, if ventilated patients actively resist lung inflation or sustain expiratory muscle activity at end-inspiration, then end-inspiratory Paw will exceed resting Paw for that lung volume. Similarly, if patient activity prevents full exhalation by expiratory braking, then for the same end-expiratory airway pressure (often measured as positive end-expiratory pressure [PEEP]), lung volume may be higher than predicted from end-expiratory Paw values alone. Finally, even if inspiration is passive and no increased airway resistance is present, Paw may rapidly increase as chest wall compliance decreases, especially as chest wall compliance includes diaphragmatic dissention. If intraabdominal pressure were to increase then end-expiratory Paw must also increase for a constant tidal volume. In acute respiratory distress syndrome (ARDS), increases in intraabdominal pressure can occur with gut wall swelling (third-spacing) and gut distention.6
As the lung expands, it pushes on surrounding structures, distorting them and causing their surface pressures to increase. Thus lung expansion induces an increase in lateral wall, diaphragmatic, and juxtacardiac Ppl as well as Ppc. The degree of increase in each of these surface pressures will be a function of the compliance and inertance of their opposing structures. These interactions were described by Novak et al.,6 who demonstrated that changes in Ppl induced by positive-pressure ventilation are not similar in all regions of the thorax and increase differently as inspiratory flow rate and frequency increase. Pleural pressure on the diaphragm increases least during inspiration, and juxtacardiac Ppl increases most. Since the diaphragm is very compliant, it seems reasonable that diaphragmatic ITP should increase less than lateral chest wall Ppl to sudden increases in lung volume. However, if abdominal distention develops, as commonly occurs in the setting of sepsis, the diaphragm will become relatively noncompliant because of the increase in abdominal pressure. Under these conditions, ITP tends to increase similarly across the thorax, so one may incorrectly assume with abdominal distention that the lung is injured and becoming stiffer. In fact, lung compliance may be normal, but chest wall compliance is restricting expansion.6 This distinction is important because increasing Paw to overcome chest wall stiffness should increase ITP more with greater hemodynamic consequences but should not improve gas exchange, since the alveoli are not damaged. If lung compliance is reduced, as in ALI, similar increases in Paw should not increase ITP as much but should also recruit collapsed and injured alveolar units, improving gas exchange but having smaller hemodynamic effects.
A hydrostatic pressure gradient exists in the pleural space. Dependent regions have a higher baseline pressure than nondependent regions in proportion to their height above or below the heart in centimeters and equate to an equal cm H2O pressure difference. In the supine subject, steady state apneic Ppl along the horizontal plane from the apex of the lung to the diaphragm are similar, whereas anterior Ppl is less, and posterior gutter Ppl is greater (Figure 47-1).
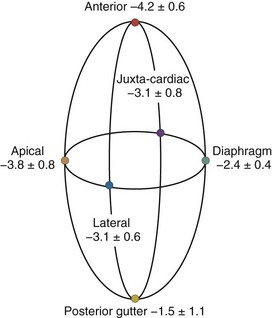
Care must be taken to determine not only what types of ventilation are being compared but also how and where estimates of Ppl and Ppc are made. For example, if estimates of transpulmonary pressure are needed to define lung compliance and its change with recruitment maneuvers, lateral chest wall Ppl appears to more accurately reflect the pressure volume characteristics of the intact lung.6 Similarly, if diaphragmatic work is to be monitored, either esophageal or diaphragmatic Ppl should be used. Finally, if heart-lung interactions are being examined, juxtacardiac Ppl is the most accurate measure of Ppl, and increases during positive-pressure inspiration will be underestimated by esophageal pressure. Since the heart is fixed within the cardiac fossa, juxtacardiac Ppl increases more than lateral chest wall or diaphragmatic Ppl. Pinsky and Guimond7 demonstrated that heart failure was associated with a greater increase in Ppc than juxtacardiac Ppl, presumably because of pericardial restraint. Importantly, with progressive increases in PEEP, juxtacardiac Ppl increased toward levels of Ppc found without PEEP, whereas Ppc initially remained constant. Once these two surface pressures became equal, further increases in PEEP increased both juxtacardiac Ppl and Ppc in parallel. If pericardial volume restraint exists, juxtacardiac Ppl will underestimate Ppc, but with sustained lung compression of the heart overriding tamponade, juxtacardiac Ppl and Ppc will become similar.
Pleural Pressure and Lung Volume in Acute Lung Injury
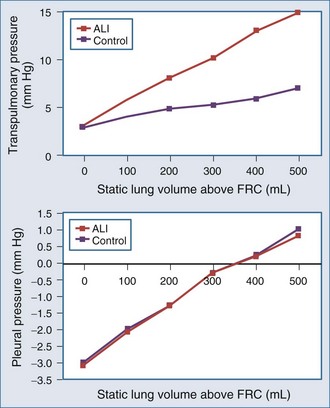
The interaction of Paw, lung volume, and ITP in the setting of lung disease is complex and can be different for the same pathologic setting depending on the tidal volume, inspiratory flow rate, ventilatory frequency, and body position. The presence of parenchymal disease, airflow obstruction, and extrapulmonary processes that directly alter chest wall–diaphragmatic contraction also profoundly alter these interactions. Static lung expansion occurs as Paw increases because the transpulmonary pressure (Paw relative to ITP) increases. If lung injury induces alveolar flooding or increased pulmonary parenchyma stiffness, greater increases in Paw will be required to distend the lungs to a constant end-inspiratory volume. Romand et al.8 demonstrated that Paw increased more during ALI than in control conditions for a constant tidal volume, whereas lateral chest wall Ppl and Ppc increased similarly between both conditions if tidal volume was held constant (Figure 47-2). These data agree with the studies of O’Quinn et al.9 that the primary determinant of the increase in Ppl and Ppc during positive-pressure ventilation is lung volume change, not Paw change. Data from Romand et al.8 demonstrated that the increase in ITP during sustained increases in lung volume is greater than the increase in Ppc. Presumably, Ppc does not increase as much as ITP because increasing lung volume also reduces filling of the ventricles, thereby reducing their size inside the cardiac fossa. To summarize, for a constant increase in lung volume, ITP will increase similarly despite drastic changes in lung compliance and airway resistance.
Relation Between Airway, Pleural, and Pericardial Pressures
Since the distribution of alveolar collapse and alterations in lung compliance in ARDS and ALI is non-homogeneous, lung distention during positive-pressure ventilation must reflect overdistention of some regions of the lung at the expense of noncompliant or poorly compliant regions. Accordingly, Paw will reflect distention of lung units that were aerated prior to inspiration but may not reflect the degree of lung inflation of nonaerated lung units. Pressure-limited ventilation assumes this is the case and aims to limit Paw in ALI so as to prevent overdistention of aerated lung units, with the understanding that tidal volume, and thus minute ventilation, must decrease. Therefore, pressure-limited ventilation will hypoventilate the lungs, leading to “permissive” hypercapnia. It is not surprising that in an animal model of ALI in which tidal volume was either kept constant at pre-injury levels or reduced to match pre-injury plateau Paw (pressure-limited ventilation), both Ppl and Ppc increased less compared to both pre–lung injury states or in ALI when tidal volume remained at pre-injury levels.8 These points underlie the fundamental hemodynamic differences seen when different modes of mechanical ventilatory support are compared to each other.
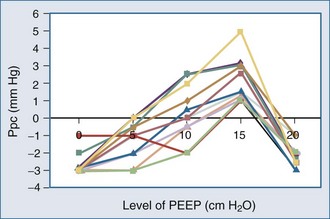
Because ALI is often non-homogeneous, with aerated areas of the lung displaying normal compliance, large increases in Paw can overdistend aerated lung units.10 Vascular structures that are distended will have a greater increase in their surrounding pressure than collapsible structures that do not distend.11 However, Romand et al.8 and Scharf and Ingram12 demonstrated that despite this non-homogeneous pattern of alveolar distention, if tidal volume is kept constant, Ppl increases in a homogeneous manner independent of the mechanical properties of the lung. Under constant tidal volume conditions, changes in peak and mean Paw will reflect changes in the mechanical properties of the lungs and patient coordination but may not reflect changes in ITP. Similarly, changes in Paw may not alter global cardiovascular dynamics. Underscoring this limitation of Paw to reflect either ITP or Ppc, Pinsky et al.13 demonstrated in postoperative patients that the percentage of Paw increase that will be transmitted to the pericardial surface is not constant from one subject to the next as PEEP is increased (Figure 47-3). Thus, one cannot predict the amount of increase in Ppc or Ppl that will occur in patients as PEEP is increased. Accordingly, assuming some constant fraction of Paw transmission to the pleural surface as a means of calculating the effect of increasing Paw on Ppl is inaccurate and potentially dangerous to patient management.
Although it may be difficult to know the actual Ppl, it is possible to determine the ventilation-induced change in Ppl. Since during airway occlusion maneuvers lung volume does not change, transpulmonary pressure is also constant so that the change in ITP is equal to the change in Paw.14 Accordingly, an increase in Paw of 20 mm Hg during a Valsalva maneuver will reflect an increase in ITP of 20 mm Hg, and a decrease in Paw to −20 mm Hg during a Mueller maneuver will reflect a decrease in ITP of 20 mm Hg below atmospheric pressure.
Clinically, esophageal pressure is often used as a surrogate for Ppc and ITP. Two limitations to the use of esophageal pressure (Pes) in estimating Ppc and ITP exist. First, Ppc and ITP may not be similar nor increase by similar amounts with the application of positive Paw if the pericardium becomes a limiting membrane.15,16 Operationally, this equates to Ppc exceeding juxtacardiac Ppl by the degree to which the pericardium limits biventricular dilation. Thus, estimates of Ppc made by using ITP measures may underestimate Ppc and overestimate the increase in Ppc as Paw is increased. Second, although esophageal pressure is often used clinically to estimate swings in both Ppl and Ppc so as to calculate the work cost of breathing, esophageal pressure is only accurate at reflecting negative swings in Ppl during spontaneous inspiration in upright seated individuals3 and in recumbent dogs in the left lateral position.17 Esophageal pressure changes underestimate both the positive swings in Ppl and the mean increase in Ppl seen with increases in lung volume during positive-pressure ventilation. During Mueller and Valsalva maneuvers, however, because lung volume does not change, swings in esophageal pressure will accurately reflect swings in ITP.3 In fact, documenting that airway and esophageal pressure swings are identical in magnitude is how esophageal manometers are validated at the bedside.
 Hemodynamic Effects of Ventilation
Hemodynamic Effects of Ventilation
Lung volume increases during both spontaneous and positive-pressure ventilation, but ITP decreases during spontaneous inspiration and increases during positive-pressure inspiration. Changes in ITP and the metabolic demand needed to create these changes represent the primary determinants of the hemodynamic differences between spontaneous and positive-pressure ventilation.18,19
Ventilation As Exercise
Spontaneous ventilatory efforts are produced by respiratory muscle contraction. Blood flow to these muscles is derived from several arterial circuits whose absolute flow is believed to exceed the highest metabolic demand of maximally exercising skeletal muscle.20 Thus, under normal cardiovascular conditions, blood flow is not the limiting factor determining maximal ventilatory effort. Although ventilation normally requires less than 5% of total O2 delivery to meet its demand,20 in lung disease states where the work of breathing is increased (e.g., pulmonary edema, bronchospasm), the work cost of breathing can increase metabolic demand for O2 to 25% or 30% of total O2 delivery.20–23 Furthermore, if cardiac output is limited, blood flow to other organs and to the respiratory muscles may be compromised, inducing both tissue hypoperfusion and lactic acidosis.24–27 Starting mechanical ventilation may reduce metabolic demand, increasing SvO2 for a constant cardiac output and CaO2. Intubation and mechanical ventilation, when adjusted to the metabolic demands of the patient, may dramatically decrease the work of breathing, resulting in increased O2 delivery to other vital organs and decreased serum lactic acid levels. These cardiovascular benefits can also be realized with effective use of noninvasive ventilation mask continuous positive airway pressure (CPAP).28 The obligatory increase in SvO2 will result in an increase in the PaO2 if fixed right-to-left shunts exist, even if mechanical ventilation does not alter the ratio of shunt blood flow to cardiac output. Finally, if cardiac output is severely limited, respiratory muscle failure develops despite high central neuronal drive such that many heart failure patients experience respiratory arrest prior to cardiovascular standstill.29
Ventilator-dependent patients who fail to wean from mechanical ventilation may occasionally have impaired baseline cardiovascular performance30 but routinely develop overt signs of heart failure during weaning, including pulmonary edema,30,31 myocardial ischemia,32–35 tachycardia, and gut ischemia.36 Jubran et al.37 demonstrated that although all subjects increase their cardiac outputs in response to a weaning trial, those who subsequently fail to wean demonstrate a reduction in mixed venous O2 saturation, consistent with a failing cardiovascular response to increased metabolic demand. Importantly, the increased work of breathing may come from endotracheal tube flow resistance.38 Thus, weaning from mechanical ventilatory support can be considered a cardiovascular stress test. Again, investigators have documented weaning-associated ECG and thallium cardiac blood flow scan-related signs of ischemia in subjects with known coronary artery disease32 and in otherwise normal patients.34,35 Placing patients with severe heart failure and/or ischemia on ventilatory support by either intubation and ventilation39 or noninvasive CPAP40 can reverse myocardial ischemia.
Hemodynamic Effects of Changes in Lung Volume
Autonomic Tone
The lungs are richly enervated with integrated somatic and autonomic fibers that originate, traverse through, and end in the thorax. These neuronal networks mediate multiple homeostatic processes through the autonomic nervous system that alter both instantaneous cardiovascular function (e.g., respiratory sinus arrhythmia) and steady state cardiovascular status (e.g., ADH-induced fluid retention). Numerous cardiovascular reflexes are centered within this network. Inflation induces immediate changes in autonomic output. The most commonly described inflation-chronotropic responses act through vagal-mediated reflex arcs.41,42 Lung inflation to normal tidal volumes (<10 mL/kg) increases heart rate via parasympathetic tone withdrawal. Inspiration-associated cardioacceleration is referred to as respiratory sinus arrhythmia (RSA)43 and denotes normal autonomic tone.44 Loss of RSA is associated with dysautonomia, and its reappearance precedes the return of peripheral autonomic control in diabetics with peripheral neuropathy.45 However, some degree of respiratory-associated heart rate change is intrinsic to the heart itself. For example, in denervated human hearts (transplants), a small degree of ventilation-associated changes in heart rate persists,46 suggesting that mechanoreceptors in the right atrium can alter sinoatrial tone. Bernardi et al.46 demonstrated that this heart rate variability in cardiac transplant recipients had a periodicity twice that of the ventilatory cycle, suggesting that both increases and decreases in venous return and ventricular loading impart some RSA. In support of this concept, Pinna et al. documented that most of the heart rate and arterial pressure changes seen during breathing in patients with severe congestive heart failure (CHF) are more reflective of changes in intrathoracic blood volume than of alterations in autonomic input.47
Lung inflation to larger tidal volumes (>15 mL/kg) decreases heart rate. Pulmonary vasoconstriction may occur through vagal reflex arcs48 but does not appear to induce significant hemodynamic effects. Reflex arterial vasodilatation can also occur with lung hyperinflation.41,49–53 This inflation-vasodilatation response appears to be mediated by afferent vagal fibers, because it is abolished by selective vagotomy. Interestingly, blocking sympathetic afferent fibers also blocks this reflex,51,54 presumably by withdrawing central sympathetic tone. Although this inflation-vasodilatation response induces expiration-associated reductions in LV contractility in healthy volunteers55 and in ventilator-dependent patients with the initiation of high-frequency ventilation41 or hyperinflation,51 its clinical significance in other patient groups is unknown. Since patients with ALI often ventilate only a relatively small amount of their lungs, the potential exists that these patients may experience regional hyperinflation and may develop reflex cardiovascular depression. Interestingly, several studies comparing larger tidal volume ventilation with pressure-limited ventilation document better hemodynamic status with pressure-limited ventilation. Similarly, although humoral factors (including compounds whose production is dependent on cyclooxygenase activation56) released from pulmonary endothelial cells during lung inflation may also induce this depressor response,57–59 these interactions do not appear to grossly alter cardiovascular status.60 In fact, unilateral lung hyperinflation (unilateral PEEP) does not appear to influence systemic hemodynamics.61 Interestingly, increased levels of nitric oxide (NO) in the exhaled gas of rabbits ventilated at increasing tidal volumes have been reported.48 Importantly, for the same decrease in cardiac output, heart rate increases less with the application of PEEP than with hemorrhage.48 The reasons for this difference are unknown but may reflect PEEP-induced sympatholytic actions and increased arterial pressure minimizing baroreceptor stimulation.
Both positive-pressure ventilation and sustained hyperinflation stimulate endocrinological responses that induce fluid retention via right atrial stretch receptors. Plasma norepinephrine, plasma renin activity,62,63 and atrial natriuretic peptide (ANP)64 increase during positive-pressure ventilation with or without PEEP. Interestingly, when subjects with CHF are given nasal CPAP, plasma ANP activity decreases in parallel with improvements in blood flow.65,66
Determinants of Pulmonary Vascular Resistance
Changes in lung volume are caused by changes in transpulmonary distending pressure, the pressure difference between alveolar pressure and ITP. Since pulmonary tissue pressure and ITP are nearly identical, increasing lung volume increases the difference between alveolar and tissue pressures, making pulmonary vascular resistance increase independent of any effect of volume change on humoral or autonomic responses.18,67–72
Lung inflation primarily affects cardiac function and cardiac output by altering right ventricle (RV) preload and afterload.72 RV afterload is the maximal RV systolic wall stress during contraction73 which, by Laplace’s law, equals the product of the RV radius of curvature (a function of end-diastolic volume) and transmural pressure (a function of systolic RV pressure).74 Changes in ITP that occur without changes in lung volume, as may occur with obstructive inspiratory efforts, will not alter the pressure gradient between the RV and pulmonary artery nor result in change of pulmonary vascular resistance. Thus, neither straining at stools (Valsalva maneuver) nor obstructive inspiratory efforts (Mueller maneuver) primarily affect RV afterload. Although obstructive inspiratory efforts are usually associated with increased RV afterload, the reason for this effect is backward LV failure and reactive hypoxic pulmonary vasoconstriction.75,76
Systolic RV pressure approximates transmural systolic pulmonary artery pressure (Ppa) when no pulmonary stenosis is present. Transmural Ppa can increase by one of two mechanisms: (1) an increase in pulmonary arterial pressure without change in pulmonary vasomotor tone, as occurs with increases in blood flow (exercise) or passive increases in outflow pressure (LV failure), or (2) an increase in pulmonary vascular resistance. Usually any increase in transmural Ppa during positive-pressure ventilation is due to an increase in pulmonary vascular resistance, because neither instantaneous cardiac output77 nor LV filling14 changes. Increases in transmural Ppa impedes RV ejection,78 decreasing RV stroke volume79 and causing RV dilation and passive obstruction to venous return,56,58 which may rapidly progress to acute cor pulmonale.80 If RV dilation and pressure overload persist, RV free wall ischemia and infarction can develop.81 Importantly, rapid fluid challenges in the setting of acute cor pulmonale can precipitate profound cardiovascular collapse due to excessive RV dilation, RV ischemia, and compromised LV filling through the process of ventricular interdependence. During normal end-inspiration, mild hypoxemia (PaO2 > 65 mm Hg) and low levels of PEEP (<7.5 cm H2O) should minimally increase transmural Ppa. If slight increases in transmural Ppa are sustained, however, fluid retention occurs, either by intrinsic humoral mechanisms (increased ANP secretion) or by therapeutic intravascular volume infusion,82 resulting in an increase in RV end-diastolic volume maintaining cardiac output.74,83
The mechanism by which ventilation alters pulmonary vasomotor tone is complex. If regional alveolar PO2 (PAO2) decreases below 60 mm Hg, local pulmonary vasomotor tone increases, reducing local blood flow.84 This process of hypoxic pulmonary vasoconstriction is mediated in part by variations in the synthesis and release of NO by pulmonary vascular endothelial cells. The pulmonary endothelium normally synthesizes a basal low amount of NO, a potent vasodilator, using endothelial nitric oxide synthase. This basal NO release is highly regulated and dependent on O2 and is inhibited by both hypoxia and acidosis. If O2 becomes scarce, NO is not made, and pulmonary vasomotor tone increases to the level that would exist if no NO were present.
Many pulmonary pathologic processes are associated with regional reductions in PAO2, such as atelectasis, airway obstruction, and ventilation/perfusion mismatching. Hypoxic pulmonary vasoconstriction, by reducing pulmonary blood flow to those hypoxic regions, optimizes ventilation/perfusion matching. However, if alveolar hypoxia occurs throughout the lungs, overall pulmonary vasomotor tone increases, raising pulmonary vascular resistance and impeding RV ejection.73 At low lung volumes, alveoli spontaneously collapse as a result of loss of interstitial traction and closure of the terminal airways, causing alveolar hypoxia. Patients with acute hypoxemic respiratory failure have small lung volumes.85,86 Therefore, pulmonary vascular resistance is often increased in these patients owing to alveolar collapse and resultant hypoxic pulmonary vasoconstriction.
Mechanical Ventilation-Induced Changes in Pulmonary Vascular Resistance
Mechanical ventilation may reduce active pulmonary vasomotor tone by one of several related processes. Hypoxic pulmonary vasoconstrictor tone may be decreased by increasing global PAO2 by enriching alveolar gas O2,87–90 reexpansion of collapsed alveolar units thereby increasing PAO2 in those local alveoli,5,91–93 increased alveolar ventilation and reversal of acute respiratory acidosis,90 or merely through decreasing central sympathetic output by allowing the patient in ALI to not fight for every breath.94,95 Similarly, these effects need not require positive-pressure breaths as much as expansion of collapsed alveoli.96 Such recruitment of lung units is usually accomplished by the addition of PEEP or CPAP. Thus, if PEEP opens collapsed lung units and replenishes alveolar gas with O2, hypoxic pulmonary vasoconstriction will be reduced, pulmonary vascular resistance will decrease, and RV ejection will improve.
Changes in lung volume can also profoundly alter pulmonary vasomotor tone by passively compressing the alveolar vessels.85,92,93 Pulmonary circulation can be separated into two groups of blood vessels depending on what pressure surrounds them92 (Figure 47-4). The small pulmonary arterioles, venules, and alveolar capillaries sense alveolar pressure as their surrounding pressure and are referred to as alveolar vessels. The large pulmonary arteries and veins, as well as the heart and intrathoracic great vessels of the systemic circulation, sense interstitial pressure or ITP as their surrounding pressure and can be called extraalveolar vessels. Alveolar pressure minus ITP is the transpulmonary pressure. Increasing lung volume requires a rise in transpulmonary pressure, so the extravascular pressure gradient between alveolar and extraalveolar vessels varies proportionally with changes in lung volume. Importantly, the radial interstitial forces of the lung that keep the airways patent91,97,98 also act upon the extraalveolar vessels. As lung volume increases, the radial interstitial forces increase, increasing the diameter of both extraalveolar vessels and airways and resulting in a reduction in airway resistance at higher lung volumes, as well as increased extraalveolar vessel diameter and capacitance.99 This tethering effect is lost with lung deflation, thereby increasing pulmonary vascular resistance.88,91 The collapse of small airways also induces alveolar hypoxia. Thus, at small lung volumes, pulmonary vascular resistance is increased owing to the combined effect of hypoxic pulmonary vasoconstriction and extraalveolar vessel collapse.
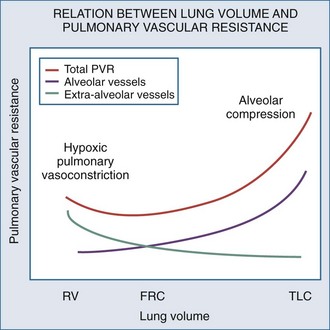
Increases in lung volume progressively raise alveolar vessel resistance, becoming noticeable above resting lung volume or functional residual capacity (FRC).88,100 There are two causes of the increased alveolar vessel resistance. First, the heart and extraalveolar vessels sense ITP as their surrounding pressure, whereas the alveolar vessels sense alveolar pressure as their surrounding pressure, so an extralumenal transpulmonary pressure gradient exists between extraalveolar and alveolar vessels. As lung volume increases, the extralumenal pressure difference increases as well. If transpulmonary pressure increases enough to exceed intralumenal vascular pressure, the pulmonary vasculature will collapse where extraalveolar vessels pass into alveolar loci, reducing the vasculature cross-sectional area and increasing pulmonary vascular resistance. Similarly, increasing lung volume by stretching and distending the alveolar septa may also compress alveolar capillaries, although this mechanism is less well substantiated. Hyperinflation can create significant pulmonary hypertension and may precipitate acute RV failure (acute cor pulmonale)101 and RV ischemia.81 Thus PEEP may increase pulmonary vascular resistance if it induces overdistention of the lung above its normal FRC. Recently the effect of inflation on RV input impedance was validated in humans, using echocardiographic techniques.102 Similarly, if lung volumes are reduced, then increasing lung volume back to baseline levels by the use of PEEP will decrease pulmonary vascular resistance by reversing hypoxic pulmonary vasoconstriction.103
Ventricular Interdependence
Changes in RV output must invariably alter LV filling because the two ventricles are serially linked through the pulmonary vasculature. However, LV preload can also be directly altered by changes in RV end-diastolic volume.104 If RV volume increases, LV diastolic compliance will decrease by the mechanism of ventricular interdependence.105 Ventricular interdependence functions through two separate processes. First, increasing RV end-diastolic volume will induce a shift of the intraventricular septum into the LV, thereby decreasing LV diastolic compliance106 (Figure 47-5). For the same LV filling pressure, RV dilation will decrease LV end-diastolic volume and therefore cardiac output. This interaction is believed to be the major determinant of the phasic changes in arterial pulse pressure and stroke volume seen in tamponade, referred to as pulsus paradoxus. Spontaneous inspiration increases venous return, causing RV dilation and decreasing LV end-diastolic compliance. Maintaining a relatively constant rate of venous return, either by volume resuscitation107 or vasopressor infusion,4 will minimize this effect. Thus, the presence of pulse paradoxus can be used as a marker of functional hypovolemia, even if actual intravascular volume status is not reduced.
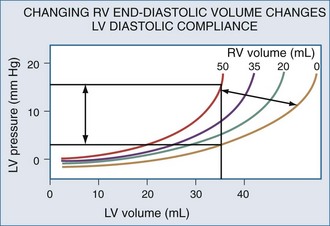
Mechanical Heart-Lung Interactions
With hyperinflation, the heart may be compressed between the two expanding lungs,108 increasing juxtacardiac ITP. The chest wall and diaphragm can move away from the expanding lungs, whereas the heart is trapped within its cardiac fossa, so juxtacardiac ITP may increase more than lateral chest wall or diaphragmatic ITP.6,16 This compressive effect of the inflated lung can be seen with either spontaneous109 or positive pressure–induced hyperinflation.97,98 This decrease in “apparent” LV diastolic compliance106 was previously misinterpreted as impaired LV contractility, because LV stroke work for a given LV end-diastolic pressure or pulmonary artery occlusion pressure is decreased.110,111 However, numerous studies have shown that when patients are fluid resuscitated to return LV end-diastolic volume to its original level, both LV stroke work and cardiac output also returned to their original levels69,107 despite the continued application of PEEP.112
Takata et al.113 proposed a novel approach to understanding tamponade that lends itself to mechanical heart-lung interactions. Hyperinflation directly alters biventricular filling, whereas inspiration primarily alters RV filling and only indirectly affects LV volumes through changes in diastolic compliance. Thus, hyperinflation—as occurs in severe asthma and with the use of excessive amounts of PEEP—would produce a clinical picture indistinguishable from tamponade. Indeed, Rebuck and Read114 made this same observation in their analysis of the hemodynamic effects of severe asthma over 30 years ago, although they did not postulate a specific mechanism to explain this phenomenon. Presumably, the shift from “uncoupled” to “coupled” cardiac fossal restraint would occur as absolute lung volume increased, biventricular volume increased, or both. If cardiac volumes are small, and lung inflation does not overdistend the chest, RV filling will be primarily impeded. In contrast, in CHF states and with marked lung overdistention, both RV and LV filling may be compromised by ventilation.
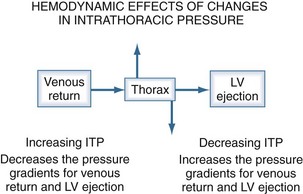
Hemodynamic Effects of Changes in Intrathoracic Pressure
The heart within the thorax is a pressure chamber within a pressure chamber. Therefore, changes in ITP will affect the pressure gradients for both systemic venous return to the RV and systemic outflow from the LV, independent of the heart itself (Figure 47-6). Increases in ITP, by increasing right atrial pressure and decreasing transmural LV systolic pressure, will reduce the pressure gradients for venous return and LV ejection, thereby decreasing intrathoracic blood volume. Using the same argument, decreases in ITP will augment venous return and impede LV ejection and increase intrathoracic blood volume.
Systemic Venous Return
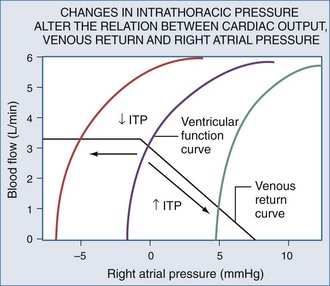
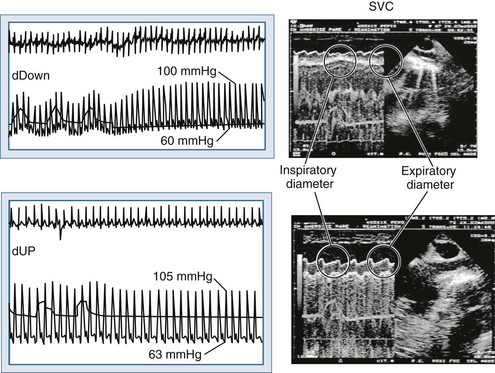
Blood flows back to the heart from the periphery through low-pressure, low-resistance venous conduits. Guyton et al. characterized venous flow from the venous reservoirs into the right atrium.115 As downstream right atrial pressure varies, as occurs with ventilation, the rate of venous return inversely changes. Pressure in the upstream venous reservoirs is called mean systemic pressure. Mean systemic pressure is a function of blood volume, peripheral vasomotor tone, and the distribution of blood within the vasculature.116 Mean systemic pressure does not change rapidly during the ventilatory cycle, whereas right atrial pressure does owing to concomitant changes in ITP. Accordingly, variations in right atrial pressure represent the major factor determining the fluctuation in pressure gradient for systemic venous return during ventilation.77,117 Positive-pressure inspiration increases ITP and right atrial pressure, decreasing the pressure gradient for venous return and RV filling,79 and consequently, RV stroke volume.77,79,118–125 These physiologic effects have recently been validated in humans, using minimally invasive echocardiographic techniques wherein vena caval flow varies with the phase of the ventilatory cycle25,126 (Figure 47-7). During normal spontaneous inspiration, the converse occurs: with decreases in ITP, right atrial pressure decreases, accelerating venous blood flow and increasing RV filling and RV stroke volume* (Figure 47-8).
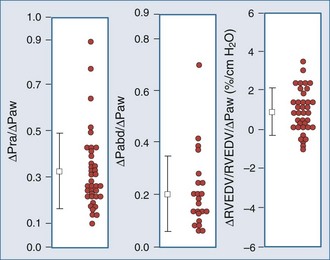
The decrease in venous return during positive-pressure ventilation is often lower than one might expect based on the increase in right atrial pressure. Fessler et al.129 and Takata and Robotham130 demonstrated in dogs that PEEP increases intraabdominal pressure by causing the diaphragm to descend, thereby increasing the pressure surrounding the intraabdominal vasculature. Because a large proportion of venous blood is in the abdomen, the net effect of PEEP is to increase mean systemic pressure and right atrial pressure. Accordingly, the pressure gradient for venous return may not be reduced by PEEP, especially in patients with hypervolemia. In fact, abdominal pressurization by diaphragmatic descent may be the major mechanism by which the decrease in venous return is minimized during positive-pressure ventilation.131–135 Furthermore, Matuschak et al.131 found that although PEEP decreased blood flow to the liver in proportion to the induced decrease in cardiac output in normovolemic dogs, the liver’s ability to clear hepatocytic-specific compounds, such as indocyanine green, was unaltered. Finally, when cardiac output is restored to pre-PEEP levels by fluid resuscitation131,136 while PEEP is maintained, liver clearance mechanisms increase above pre-PEEP levels.136–139 These data are consistent with a PEEP-induced alteration in intrahepatic blood flow distribution. Thus, ventilation may have less of an effect on venous return than originally postulated, but the effect may be more complicated than we have imagined. Van den Berg et al.140 examined the effects of varying levels of CPAP on right atrial pressure, intraabdominal pressure, and cardiac output in 42 postoperative cardiac surgery patients. Up to 20 cm H2O CPAP did not significantly decrease cardiac output, as measured 30 seconds into an inspiratory hold maneuver. The reason for this apparent paradoxical effect became obvious when they compared the associated changes in right atrial pressure, abdominal pressure, and RV end-diastolic volume (Figure 47-9). What they documented was that only 30% of the increased airway pressure was transmitted to the right atrium. Perhaps more importantly, most of the increase in right atrial pressure was also realized by an increase in intraabdominal pressure, so it was not surprising that RV end-diastolic volume fell by less than 8% from pre-CPAP values. These data demonstrate that in the fluid-resuscitated patient, institution of positive-pressure ventilation may not result in a decrease in blood flow. However, if intraabdominal pressure is allowed to decrease, as would occur with an open laparotomy and decompression of tense ascites, a marked preload-responsive effect of positive-pressure ventilation can occur.
With exaggerated swings in ITP, as occur with obstructed inspiratory efforts, venous return behaves as if abdominal pressure is additive to mean systemic pressure in defining total venous blood flow.141–144 Recent interest in inverse ratio ventilation has raised questions as to its hemodynamic effect because its application includes a large component of hyperinflation. However, Mang et al.145 demonstrated in an animal model of ALI that if total PEEP (intrinsic PEEP plus extra extrinsic PEEP) was similar, no hemodynamic difference between conventional ventilation and inverse ratio ventilation was seen.
Right Ventricular Filling
Under normal conditions, it is difficult to document any relation between RV filling pressure and volume. When RV filling pressure, defined as right atrial pressure minus Ppc, was directly measured in patients undergoing open chest operations, RV filling pressure was unaltered by acute volume loading.146 Although right atrial pressure increased, Ppc also increased such that RV filling pressure remained unchanged. Similar data were seen when RV volumes are reduced by the application of PEEP in postoperative cardiac patients.147 These findings suggest that under normal conditions, RV diastolic compliance is very high, and most of the increase in right atrial pressure seen during volume loading reflects pericardial compliance and cardiac fossa stiffness, more than changes in RV distending pressure. These data also imply that with RV filling, right heart sarcomere length remains constant. Presumably, conformational changes in the RV more than wall stretch are responsible for RV enlargement.15 Accordingly, changes in right atrial pressure do not follow changes in RV end-diastolic volume. When cardiac contractility is reduced and intravascular volume is expanded, RV filling pressure increases as a result of decreased RV diastolic compliance, increased pericardial compliance, increased end-diastolic volume, or a combination of all three. In support of this hypothesis, RV filling pressure does not increase until RV volume exceeds a certain threshold value.105 Furthermore, in dogs with acute ventricular failure, volume loading increases Ppc more than ITP, consistent with pericardial rather than cardiac fossal restraint. If PEEP is increased in this setting, ITP but not Ppc selectively increases until ITP equals Ppc, then both ITP and Ppc increase equally if PEEP is increased further.7 In postoperative cardiac surgery patients,13,22,148 PEEP—and by extension lung expansion—compresses the heart within the cardiac fossa in a fashion analogous to pericardial tamponade.
Venous return is the primary determinant of cardiac output.116 Since right atrial pressure is the backpressure to venous return, venous return is maintained near maximal levels at rest51,125,126 because RV filling occurs with minimal changes in filling pressure.148 The closer right atrial pressure remains to zero relative to atmospheric pressure, the greater the pressure gradient for systemic venous blood flow.115,121 For this mechanism to operate efficiently, RV output must equal venous return, otherwise sustained increases in venous blood flow would overdistend the RV, increasing right atrial pressure. Fortunately, under normal conditions of spontaneous ventilation, the increase in venous return is in phase with inspiration, decreasing again during expiration as ITP increases.77 Likewise, the pulmonary arterial inflow circuit is highly compliant and can accept large increases in RV stroke volume without changing pressure.79,83 Thus, any increase in venous return is proportionally delivered to the pulmonary circuit without forcing the RV to increase its force of contraction or myocardial oxygen demand. Importantly, this compensatory system will rapidly become dysfunctional if RV diastolic compliance decreases or if right atrial pressure increases independent of changes in RV end-diastolic volume. An example of decreased RV diastolic compliance is acute RV dilation or cor pulmonale (pulmonary embolism, hyperinflation, and RV infarction) that induce profound decreases in cardiac output not responsive to fluid resuscitation. Dissociation between right atrial pressure and RV end-diastolic volume occurs during either tamponade or positive-pressure ventilation, because right atrial pressure is artificially increased by the increasing ITP. Accordingly, positive-pressure ventilation impairs normal circulatory adaptive processes. Furthermore, even if one restores the coupling of right atrial pressure and RV volume by using partial ventilatory support modes of ventilation, cardiac output will increase only if the RV can transduce the associated increase in venous return to forward blood flow. Thus, during weaning from mechanical ventilation, occult RV failure may be exposed and will manifest as a rapid rise in right atrial pressure and a fall in cardiac output. Since the primary effect of any form of ventilation on cardiovascular function in normal subjects is to alter RV preload via altering venous blood flow, the detrimental effect of positive-pressure ventilation on cardiac output can be minimized by either fluid resuscitation to increase mean systemic pressure4,118,140,141 or by keeping both mean ITP and swings in lung volume as low as possible. Accordingly, prolonging expiratory time, decreasing tidal volume, and avoiding PEEP all minimize the decrease in systemic venous return to the RV.1,21,77,120–124,149
Since spontaneous inspiratory efforts increase lung volume by decreasing ITP, one sees an increase in venous return with spontaneous inspiration owing to the fall in right atrial pressure.19,67,121–123 However, this augmentation of venous return is limited142,143 because if ITP decreases below atmospheric pressure, venous return becomes flow-limited as the large systemic veins collapse as they enter the thorax.115 This flow limitation is a safety valve for the heart because ITP can decrease greatly with obstructive inspiratory efforts,52 and if not flow-limited, the RV could become overdistended and fail150 (see Figure 47-8). Still, in patients with decreased RV compliance, negative swings in ITP can augment RV filling. Interestingly, negative pressure ventilation, by augmenting venous return, was shown to increase cardiac output by 39% in intubated children following repair of tetralogy of Fallot.151 In this condition, impaired RV filling secondary to RV hypertrophy and reduced RV chamber size are the primary factors limiting cardiac output.


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


