Fig. 3.1
Schematic of sample processing to gene chip analysis. A schematic of the methodology involved to process an experimental tissue, hybridize the target cDNA to the gene chip, and then analyze the chip for meaningful genomic expression values
In addition to high-throughput genomic analysis, another complimentary tool for the analysis of proteins and other physiologic biomarkers is based on multiplex technologies intended to detect low levels of multiple protein types from a single tissue, blood, or urine sample. Multiplex technology is bead-based multiplexing, where beads are internally dyed with fluorescent dyes to produce a specific spectral or fluorescence address. Biomolecules, such as antibodies, can be conjugated to the surface of inert beads to capture physiologic molecules of interest. This technology uses flow cytometric or other imaging technologies for characterization of the beads as well as detection of the light emission due to target molecule presence. Luminex™ (Austin, TX) technology enables up to 500 proteins to be detected in each well of a 96-well plate, using very small starting sample volumes. Common applications include cytokines, chemokines, metabolic markers, autocrines, and phosphorylated proteins. A schematic representation of the tissue processing through data readout using a multiplex approach to cytokine analysis is depicted in Fig. 3.2.
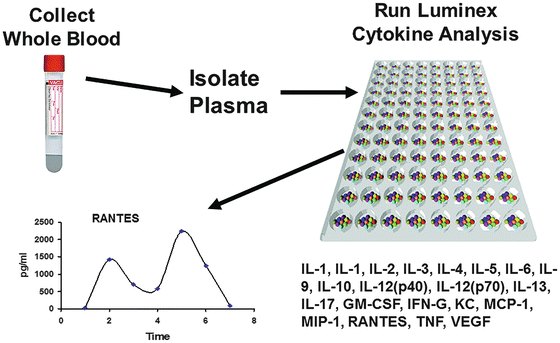
Fig. 3.2
Multiplex cytokine analysis. A schematic representation of the tissue processing through data readout required using a multiplex approach to cytokine analysis
Affymetrix GeneChip® Arrays (Santa Clara, CA)
Affymetrix makes quartz chips for genome-wide analysis called GeneChips. Affymetrix’s GeneChips enable researchers to quickly scan for the presence of particular genes in a variety of biological samples. Moreover, Affymetrix is uniquely focused on oligonucleotide microarray technology. These specific microarrays are incorporated to determine which genes exist in a biologic sample by detecting precise pieces of mRNA.
Each gene on an Affymetrix microarray GeneChip is typically represented by a probe set consisting of 11 different pairs of 25-bp oligos covering features of the transcribed region of that gene. Each pair consists of a perfect match (PM) and a mismatch (MM) oligonucleotide. The PM probe exactly matches the sequence of a particular standard genotype, often one parent of a cross, while the MM differs in a single substitution in the central, 13th base. The MM probe is designed to distinguish noise caused by nonspecific hybridization from the specific hybridization signal. Affymetrix GeneChip microarrays are the most popular high density oligonucleotide gene expression arrays and have become an invaluable tool in genomics studies worldwide. Figure 3.3 represents an actual Affymetrix Human Gene Chip U133 plus 2.0.

Fig. 3.3
Affymetrix U133 2.0. This is an actual Affymetrix U133 2.0 human GeneChip®. A single gene chip packed with fragments from each of the 35,000 known genes that make up the human genome. (Courtesy of Affymetrix, Inc., Santa Clara, CA, USA)
Microarray Technology Impacts Surgical Care
Each human cell contains approximately three billion DNA base pairs that are estimated to encode approximately 30,000 genes that are responsible for maintaining the structural and functional integrity of a cell. These genes encode the RNA and proteins that produce the cell phenotype. A neoplastic phenotype develops out of an aberration or alteration in the normal expression of genes. A variety of specific mechanisms have been implicated in the transformation process including chromosomal rearrangement, deletions, amplification, methylation, and mutations of genes. Elucidating the fundamental molecular mechanisms that are involved in the stepwise progression from normal tissues to malignant tumors, organ-specific inflammation or systemic inflammation is essential in our knowledge of the development of cancers. This would ultimately lead to improved methods of detection, treatment, and cures for cancers. Comprehensive analyses of cancer genomes promise to better inform prognoses and improve precise cancer treatments improving patient-centered and specific cancer care and chemotherapeutics [1].
Melanoma
Malignant melanoma is a highly aggressive disease accounting for a majority of the deaths from skin malignancies with patient survival dependent on early detection and diagnosis. Although some promising new therapies have recently emerged, a better understanding of the molecular alterations involved in melanoma progression, particularly from localized tumors to metastasis, such as genomic and epigenetic aberrations, will aid in early detection and development of biomarkers and future targeted treatment strategies.
Melanoma, like other solid tumors, is thought to arise from a series of genetic and epigenetic events. Genetic aberrations have been identified in the past decade and have potential utility as biomarkers. Multiple studies have revealed that epigenetic events, such as genomic promoter region methylation of CpG islands, histone modification, and microRNA (miRNA) expression, have been shown to be important regulators of melanoma progression, and that these epigenetic changes can potentially serve as molecular biomarkers in tumor tissues and in blood as circulating DNA, for diagnosing disease and predicting disease outcome and progression [2].
Epigenetics refers to heritable changes in gene expression that are not caused by changes in the genomic DNA sequence. DNA methylation is one of the hallmark epigenetic events most studied in cancers. DNA methylation involves the addition of a methyl group to the 5′ carbon of a cytosine ring located 5′ to a guanosine base in a CpG dinucleotide and is catalyzed by DNA methyltransferases. Methylation events of promoter regions have been strongly implicated in cutaneous melanoma progression. Many of these genes are involved in cell cycle control, cell signaling, migration and invasion, apoptosis, angiogenesis, and metastasis. Comparing the methylation status of melanoma primary tumor and metastasis, an increase in hypermethylation of WIF1, TFPI2, RASSF1A, and SOCS1 was seen with increasing clinical tumor stage. Despite advances in this field and development of multiple platforms for studying genomic methylation, uniformity and standardization remain significant issues in evaluating and comparing results.
miRNAs are evolutionarily conserved, endogenous, noncoding RNA transcripts of 22 nucleotides in length that serve to temporally and spatially regulate biological function. miRNAs are considered an epigenomic mechanism that can have normal regulatory function but also can have negative influence when dysregulated, particularly in cancer progression as in melanoma. They are derived from noncoding intergenic or intronic regions of DNA that, once in their mature form, interfere with protein translation from mRNA transcripts. miRNA can preferentially bind with mRNA transcripts to inhibit translation or degrade the mRNA transcript before translation can take place. miRNA can modulate biological functions such as cell cycle, proliferation, apoptosis, and angiogenesis, which, if aberrantly regulated, can lead to malignancy. As an example miR-221/222 was found to interfere with c-KIT and p27, causing dysregulation of the cell cycle during the progression of melanoma. miRNA regulation may influence the microenvironment and can contribute to tumor cell invasion, migration, and metastasis. miRNA regulation of protein coding genes is, therefore, an essential regulatory element in biological development and function [2]. Lastly, deregulated miRNA expression may serve as diagnostic or prognostic biomarker in cutaneous melanoma.
By collectively assimilating the ever evolving cadre of miRNA, biomarkers, epigenetic phenomena, and small molecule expression markers, genomic expression finger prints can be developed that serve to stratify malignant melanoma metastatic risk, responsiveness to therapy, and even long-term survival odds prospectively and prior to therapeutic intervention. No more will the responsiveness to therapy be the clinicians best guess and experience but rather based on individualized genetic and biomarker-specific expression matrix gathered from each patient.
Prostate Cancer
Prostate cancer is the most common type of cancer in men and the second leading cause of cancer death in men in the United States. The recent surge of high-throughput sequencing of cancer genomes has supported an expanding molecular classification of prostate cancer. Translation of these basic science studies into clinically valuable biomarkers for diagnosis, prognosis, and biomarkers that are predictive for therapy is critical to the development of precision medicine in prostate cancer. Many recent genomic applications are aimed at improving screening specificity in prostate cancer by differentiating aggressive versus indolent prostate cancers. Over the past decade a host of candidate gene biomarkers have been centered on three distinct groups involving ETS gene rearrangements, PTEN inactivation, and androgen receptor signaling. These and other putative biomarkers may provide a rationale for matching patients with molecularly targeted therapies in clinical trials [3].
One of the major barriers prohibiting scientist, oncologist, and clinicians from better understanding comprehensive analyses of prostate cancer genomes is the inaccessibility of metastatic tissue. A potential solution is to characterize circulating prostate tumor cells (CTCs). In the past this has required overcoming the challenges of isolating rare and metastatic prostate cancer cells and sequencing low-input material. However, recently investigators have reported an integrated process to isolate, qualify, and sequence whole exomes of prostate CTCs with high fidelity using a census-based sequencing strategy. The authors’ data support the notion that CTC sequencing can reveal early mutations in tumor evolution and those that could be shared among metastatic sites. The genomic technology involved provides a minimally invasive window into the mutational landscape of metastatic prostate cancer and will provide investigators and clinicians with more robust tools to better tailor prostate cancer chemotherapeutics in cases of metastatic disease [4].
Breast Cancer
Cancer genomics has already revolutionized our knowledge of breast cancer molecular pathology, fostering the development of new and more effective clinical protocols. The most recent advances in the field of cancer genomics and epigenomics include DNA alterations and driver gene mutations, gene fusions, DNA methylation, and miRNA expression [5].
Genome-wide association studies have allowed the discovery of only a portion of genomic alterations associated with hereditary breast cancer risk. The genes BRCA1, BRCA2, TP53, PTEN, STK11, and CDH1 with high-penetrance mutations account for only 20–25 % of all cases of hereditary breast cancer. It has been speculated that the majority of breast cancer may be explained by the accumulation of low-penetrance and acquired genetic alterations, interacting together in a polygenic model, associated with a modification of the epigenetic component. The exact definition of the patient-specific occurrence and interaction of genomic alteration is expected to be the greatest contribution that next generation sequencing will provide to breast cancer management. Until the identification of these low-penetrance genomic influences is completed, treatment of breast cancer will continue to depend on the level of expression of receptors for estrogen, progesterone, and epidermal growth factor type 2 receptor (HER2/neu) [5]. However as the genomic expression profiles evolve new chemotherapeutics and chemotherapeutic combinations will be tailored to treat individual patients with breast cancer.
Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) affects over 2.5 million people of European ancestry, with a rising prevalence in other countries. There is overwhelming evidence for the role of genetics in IBD as evidenced by initial reports of familial clustering. The last 15 years have seen a tremendous degree of progress regarding the identification of genetic loci involved in IBD. This has happened in part because of technological advances and growth in the genetic approaches used to identify these genes. Although other variations do exist, Crohn’s disease and ulcerative colitis are the two most common forms of inflammatory disease and continue to confound investigators and clinicians [6].
Genome-wide association studies and meta-analyses of patients with these two diseases have implicated a sundry of previously unsuspected cellular mechanisms, such as autophagy, in their pathogenesis. Furthermore these findings have shown that some IBD loci are shared with other inflammatory diseases like ankylosing spondylitis. Meta-analysis of Crohn’s disease and ulcerative colitis combined a total of more than 75,000 cases and controls in genome-wide association scans. The investigators found 71 new genomic associations for a total of 163 IBD loci that met genome-wide significance thresholds. The authors found that most of the discovered loci contribute to both IBD phenotypes. In addition many of the IBD-associated loci were also implicated in other immune-mediated disorders such as ankylosing spondylitis and psoriasis. An additional and unexpected finding was that there is considerable overlap between susceptibility loci for IBD and mycobacterial infection. Gene co-expression canonical pathway and network analysis emphasize this relationship, with pathways shared between host responses to mycobacteria and those predisposing to the development of IBD [7]. The findings suggest that either mycobacteria directly or the immune processes involved in mycobacteria eradication cause immune dysregulation which potentiates IBD development.
As a consequence of the ability to simultaneously scan and catalog thousands of genomic expression patterns for a disease state, biomarker panels with a high level of diagnostic sensitivity and specificity have been developed to more quickly and accurately identify patients with IBD. One such panel used in the diagnosis of Crohn’s disease is the PROMETHEUS® Crohn’s Prognostic test (Prometheus Labs, San Diego, CA). This technology combines proprietary serologic and genetic biomarkers in a logistic regression model to provide individual patients with probabilities for developing disease complications after diagnosis with Crohn’s disease. The test enables physicians to stratify patients according to their risks of developing complications and develop patient-centered treatment plans rather than a blanket therapeutic approach.
Microarray Technology in Trauma and Critical Care
The Inflammation and the Host Response to Injury collaboration is a large-scale research program centered on the immune-inflammatory response in trauma and sepsis. The mission of the multi-institutional collaborative effort funded by the National Institute of Health is to understand key regulatory elements that drive the host’s response to traumatic injury and its accompanying systemic inflammation response. The participating investigators used a discovery-driven approach to acquire large amounts of new human genomic data to try and identify genomic expression patterns that may serve as new targets for therapeutic development. In an effort to streamline the clinical sample processing and analysis, the investigators were able to develop novel genome-wide microarray technologies and introduce high-throughput proteomics to clinical medicine. These efforts have culminated in the identification of novel genomic and proteomic markers that predict patient outcomes and serve as new therapeutic targets for basic and clinical research and pharmaceutical development.
Related posts:
 End Points of Resuscitation
End Points of Resuscitation
 Cardiac Surgery Advances: Do We Still Remember How to Do the Open Bypass?
Cardiac Surgery Advances: Do We Still Remember How to Do the Open Bypass?
 The Role of Robotics in Selective Thoracic Surgical Problems: Technical Considerations
The Role of Robotics in Selective Thoracic Surgical Problems: Technical Considerations
 Artificial Limbs for Upper Extremity Amputation
Artificial Limbs for Upper Extremity Amputation
 Use of Biologic Grafts in Surgery
Use of Biologic Grafts in Surgery
 Brain Cancer: The New Frontiers
Brain Cancer: The New Frontiers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


