INTRODUCTION
Circulatory shock has a high mortality. Severe hemorrhage after injury carries a mortality rate of 30% to 40% and is responsible for almost 50% of deaths occurring within 24 hours of injury.1,2 Septic shock has a mortality of up to 50%.3 Resuscitation, starting in the prehospital setting and continuing throughout the victim’s care in the ED and on into the hospital, has the goal of restoring the necessary level of tissue perfusion and oxygenation for survival while simultaneously limiting further volume loss.
Intravascular volume depletion is a common feature of circulatory shock; crystalloids, colloids, and blood products (packed red blood cells and plasma) are the primary volume expanders to reverse intravascular volume depletion. This chapter focuses on the issues related to fluid and blood resuscitation in traumatic shock, with an emphasis on hemorrhagic shock. Processes that cause loss of plasma fluid and electrolytes (e.g., dehydration, burns, sepsis), often requiring aggressive fluid therapy, are discussed in other chapters (see chapters 150, “Toxic Shock Syndromes,” 151, “Sepsis,” and 216, “Thermal Burns”).
The principal objectives of fluid and blood resuscitation in traumatic shock are: (1) to restore intravascular volume sufficient to maintain oxygen-carrying capacity and tissue perfusion for adequate cellular oxygen delivery, and (2) to prevent or correct derangements in coagulation.
PATHOPHYSIOLOGY
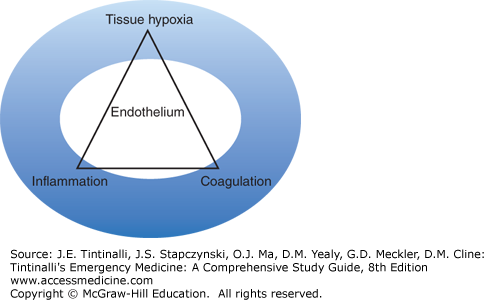
Circulatory shock is a state of impaired oxidative metabolism and homeostasis due to inadequate oxygen delivery to meet metabolic demand, and hypoperfusion leading to inadequate cellular waste removal. Acute shock triggers a complex range of physiologic responses that compensate for intravascular volume loss and maintain perfusion to the most important vascular beds. Shock also produces a global insult to the vascular endothelium that activates the coagulation and inflammatory systems (Figure 13-1). When uncorrected, coagulopathy, additional inflammation, and organ system damage result.
While moderate transient hypoperfusion may be well tolerated, prolonged or severe hypoperfusion leads to accumulation of oxygen debt and progressive cellular and organ dysfunction. If rapid and severe, sudden cardiovascular collapse and death may occur.
Coagulopathy observed in trauma victims, termed trauma-induced coagulopathy, is ascribed to a combination of factors beginning with loss of coagulation factors from hemorrhage, followed by hemodilution from crystalloid resuscitation, and then exacerbated by acidosis (evidenced by a base deficit) and hypothermia that occur during the course of ongoing hemorrhage and resuscitation.4 These same principles also apply to other causes of shock.5 However, acidosis does not, by itself, have a significant effect on coagulation until the pH decreases below 7.0.6
The lethal triad of hypothermia, hemodilution, and acidosis can be viewed a partially iatrogenic based on the type of resuscitation provided. The normal total circulating volume of an adult is approximately 7% of ideal body weight or about 5 L for an average 70-kg adult patient divided into about 3 L of plasma and 2 L of red blood cell volume (Table 13-1). Understanding this distribution explains how trauma-induced coagulopathy can be iatrogenically produced.
| Fluid Compartment | % Body Weight* | % Body Water* | Volume* (70 kg) |
|---|---|---|---|
| Total body water | 60 | 100 | 42 L |
| Intracellular | 40 | 67 | 28 L |
| Extracellular | 20 | 33 | 14 L |
| Interstitial | 15 | 25 | 10 L |
| Intravascular | ~7–8 | ~11–13 | ~5.0–5.5 L |
| Plasma | ~4.0–4.5 | ~7 | ~3.0–3.5 L |
| Red blood cells† | ~3.0–3.5 | ~5 | ~2.0–2.5 L |
Factors exacerbating trauma-induced coagulopathy, including hyperfibrinolysis and clotting factor and platelet consumption or dysfunction, are related to the degree of injury and hemorrhage.7 Early trauma-induced coagulopathy is characterized by anticoagulation and hyperfibrinolysis, likely modulated through the protein C pathway. Endothelial damage associated with trauma or sepsis stimulates an increase in thrombomodulin expression on the endothelium that complexes with thrombin and in turn activates protein C (Figure 13-2). Complexed thrombin is now no longer available for its usual hemostatic role of cleaving fibrinogen to form fibrin. Subsequent consumption of plasminogen activator inhibitor-1 by activated protein C contributes to hyperfibrinolysis (Figure 13-2).8
FIGURE 13-2.
Endothelial damage results in increased thrombomodulin (TM) expression. TM can complex with thrombin (thrombin-TM complex), leading to activation of the protein C (PtnC to aPntcC) pathway. Modulated through a multistep pathway leading to plasminogen activator inhibitor-1 (PAI-1), fibrinolysis is encouraged, resulting in increased fibrin degradation products (FDPs). In addition, the complexed thrombin is no longer able to perform its usual role in cleaving fibrinogen to fibrin to form clots.
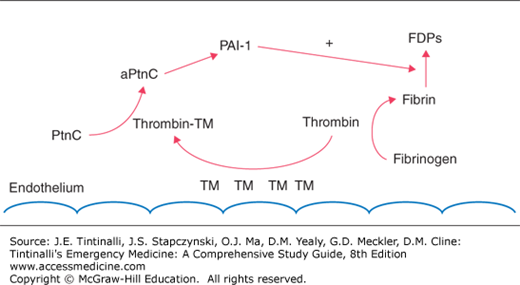
Thrombomodulin normally found in the endothelium may be viewed as protective because it is able to sequester thrombin and thus allow for generation of adequate levels of protein C to prevent thrombosis caused by low-flow states. However, on a mass systemic level, this appropriate local response may become pathologic, resulting in clotting inhibition.
CLINICAL FEATURES
The clinical appearance of acute traumatic shock is variable and depends on the cause, rate, volume, and duration of volume loss or bleeding; the presence of other acute disorders; the effects of current medications; and the patient’s baseline physiologic status.
The hemodynamic response to acute severe hemorrhage-induced hypovolemia traditionally includes tachycardia, hypotension, and signs of poor peripheral perfusion (cool, pale, clammy extremities with weak peripheral pulses and prolonged capillary refill). Arterial and venous vasoconstriction leads to a narrowing of the pulse pressure. Cerebral hypoperfusion causes alterations in mental status. With increasing blood loss, signs and symptoms become more pronounced. Classification of hemorrhage severity as a percentage of blood volume loss based on vital signs is not accurate and should not be used to guide ED resuscitation.9
Patients with excellent baseline physiologic status (e.g., young athletes) may have such a robust compensatory response to hemorrhage that they appear stable and do not manifest tachycardia or hypotension, even with large blood volume loss. Signs of peripheral hypoperfusion and subtle mental status alterations may be the only clues that the severity of hemorrhage is greater than predicted based on hemodynamic parameters. Elderly patients may not develop tachycardia due to underlying heart disease or medications such as β-adrenergic blockers. Bradycardia or lack of tachycardia may occur in about 30% of patients with intra-abdominal hemorrhage from increased vagal tone in response to hemoperitoneum. In a pregnant trauma patient, compression of the inferior vena cava by the gravid uterus can decrease central venous return and worsen hypotension and tachycardia in the setting of less severe hemorrhage.
DIAGNOSIS
Vital signs offer little value unless they are in extreme low ranges. Arterial blood pressure does not adequately reflect cardiac output or regional perfusion. Clinical evidence of peripheral hypoperfusion is useful but is not a quantitative measurement. Metabolic information (discussed below), assessment of mechanisms of injury and illness, and appropriate imaging studies offer the best chance for early recognition of severe hemorrhage and for guiding an appropriate response to treatment.
There is a biphasic relationship between oxygen consumption and oxygen delivery (Figure 13-3). While this relationship exists for the body as a whole, it also exists for each individual organ system and is largely mediated by the microcirculation. Oxygen debt develops in tissues or organs when oxygen delivery does not meet the metabolic demands and represents the amount of extra oxygen that is needed to metabolize the accumulated products of anaerobic metabolism once perfusion is restored. Oxygen debt is the accumulation of multiple oxygen deficits over time and is a measure of whole-body ischemia. Oxygen debt is the only physiologic measure that has clearly been linked to both mortality and even morbidity in the form of multiple organ failure after shock. The degree of oxygen debt incurred after injury has also been clearly linked to inflammation.10
FIGURE 13-3.
The biphasic relationship of oxygen delivery (Do2) and oxygen consumption (Vo2). As Do2 decreases, Vo2 may remain constant due to an increase in the ratio of extracted oxygen (OER) at the tissue level. This is mirrored by a decrease in venous hemoglobin oxygen saturation (Svo2). However, at some point, OER will not meet the Vo2 demands of the tissues, resulting in a state of Do2 dependent Vo2 whereby aerobic Vo2 transitions to anaerobic Vo2. At this point of critical Do2, oxygen debt begins to accumulate along with metabolic byproducts of anaerobiosis such as lactate. While this relationship exists for the body as a whole, it also exists for each individual organ system and is largely mediated by the microcirculation. [©Kevin R. Ward, MD.]

Measurements of lactate and base deficit are in fairly ubiquitous use in trauma centers and EDs and are now even being used as point-of-care testing in the prehospital setting as a triage tool. Using lactate and/or base deficit as a resuscitation trigger and its clearance/normalization as an endpoint is prudent and is associated with improvements in survival. However, elevated lactate and base deficit is a late finding, which is first preceded by increases in oxygen extraction as indicated by a decrease in indicators of oxygen consumption such as mixed or central venous hemoglobin oxygen saturation (Figure 13-2). Furthermore, improvements in oxygen delivery resulting in lactate clearance do not indicate that oxygen extraction is normal.
Although lactate is also a measure of oxygen debt, its resolution during resuscitation does not ensure the important repayment and resolution of oxygen debt. Lactate also suffers from the intermittent and time-dependent nature of its measure and change. To counter these, continuous measures of oxygen consumption surrogates are available in the form of invasive oximetric catheters measuring central venous oxygen saturation (ScvO2) and tissue oxygen saturation (StO2) noninvasively from target tissues such as skeletal muscle using near-infrared spectroscopy. StO2 measurements with infrared spectroscopy are based on the fact that tissue blood volume is 70% to 80% venous blood with the remaining 10% to 20% a combination of arterial and capillary blood. Thus aggregate measures of hemoglobin oxygenation in the tissue are heavily dominated by the venous blood and thus the postextraction compartment. Although not an exact mirror of ScvO2, ensuring StO2 values above 50% appears to be reasonable from both a diagnostic and treatment standpoint to begin and sustain hemostatic resuscitation. A target StO2 or ScvO2 between 60% and 70% should be maintained to help ensure resolution of tissue hypoxia.
TREATMENT
Resuscitation begins in the prehospital setting and continues in the ED. The priority for prehospital care is treatment of life-threatening conditions and rapid transport to an appropriate facility. For the hemorrhaging patient, this entails assuring adequate ventilation and oxygenation (including securing an airway if necessary), controlling external bleeding (if present), and protecting the spinal cord (if potentially vulnerable).
In the ED, restore intravascular volume to reverse or limit systemic and regional hypoperfusion, maintain oxygen-carrying capacity so that tissue oxygen delivery meets demand, and limit ongoing blood loss and prevent the development of coagulopathy. From the moment resuscitation begins, prevent the development of hypothermia by keeping the patient warm and administering warmed IV fluids and blood products.11
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


