Chapter 101 Endotheliopathy
Until recently, scientists and clinicians considered the endothelium, the cell layer that lines the blood vessels, as an inert barrier separating the various components of blood and the surrounding tissues. The vascular endothelium is now recognized as a highly specialized and metabolically active organ performing a number of critical physiologic, immunologic, and synthetic functions. These functions include regulation of vascular permeability, fluid and solute exchange between the blood and interstitial space, vascular tone, cell adhesion, homostasis, and vasculogenesis.1
The endothelium lies between the lumen and the vascular smooth muscle, where it is uniquely positioned to “sense” changes in hemodynamic forces or blood-borne signals by membrane receptor mechanisms. The endothelial cells can respond to physical and chemical stimuli by synthesis or release of a variety of vasoactive and thromboregulatory molecules and growth factors. Substances released by the endothelium include prostacyclin, NO, endothelins, endothelial cell growth factors, interleukins, plasminogen inhibitors, and von Willebrand factor (vWF). The vascular endothelium possesses numerous enzymes, receptors, and transduction molecules, and it interacts with other vessel wall constituents and circulating blood cells. In addition to these universal functions, the endothelium may have organ-specific roles that are differentiated for various parts of the body, such as gas exchange in the lungs, control of myocardial function in the heart, or phagocytosis in the liver and spleen. From a structural perspective, endothelial cells from different sites of the vascular tree differ in size, shape, thickness, and nuclear orientation. For example, endothelial cells that line the pulmonary artery of the rat are larger and more rectangular than those lining the aorta, whereas endothelial cells in the aorta are thicker than their counterparts in capillaries or veins.2
Normal Endothelial Function
Endothelial Cell Heterogeneity
Many vascular diseases appear to be restricted to specific vascular beds. For example, thrombotic events are often localized to single vessels. It is also common for certain vasculitides to specifically affect certain arteries, veins, or capillaries, or to affect certain organs. Tumor cells will often metastasize more commonly within particular vascular beds. The basis for this variability in vascular disease is poorly understood, but may be explained in the heterogeneity of endothelial cells. Recently, there has been a greater understanding of how endothelial cell heterogeneity may contribute both to the maintenance of organ-specific function and to the development of disorders restricted to specific vascular beds.1–3
The morphological appearance of capillary endothelium from different vascular beds may explain differences in tissue function. For example, the brain microcirculation is lined by endothelial cells connected by tight junctions that maintain the blood-brain barrier. By contrast, sinusoids found in the liver, spleen, and bone marrow are lined by endothelial cells that allow transcellular trafficking between intercellular gaps. Similarly, fenestrated endothelial cells found in the intestinal villi, endocrine glands, and kidneys facilitate selective permeability, which is required for efficient absorption, secretion, and filtering. 4
Another example of endothelial cell heterogeneity lies in the expression of cell surface receptors involved in cell-to-cell signaling and cell trafficking. For example, in the mouse, lung-specific endothelial cell adhesion molecules are exclusively expressed by pulmonary postcapillary endothelial cells and some splenic venules. Similarly, specific mucosal cell adhesion molecules are expressed primarily on endothelial venules in the Peyer patches of the small intestine.5,6 Tumor cells may show clear preferential adhesion to the endothelium of specific organs paralleling their in vivo metastatic propensities.7 Distinct subsets of endothelial cells often exist within a single organ. In in situ studies, two distinct sinusoidal endothelial cell phenotypes can be recognized in the adult human liver: hepatic periportal vessels express specific cell surface molecules such as PECAM-1 and CD34, whereas sinusoidal intrahepatic endothelial cells do not.
Endothelial Progenitor Cells
EPCs are a specific subtype of hematopoietic stem cells that have been isolated from circulating mononuclear cells, bone marrow, and cord blood. EPCs migrate from the bone marrow to the peripheral circulation, where they contribute to vascular repair.8,9 When injected into animal models of ischemia, EPCs are incorporated into sites of neovascularization,8,10,11 and have contributed to improved outcomes in patients with ischemic vascular disorders.12 In addition, there has been accumulating evidence for the function of EPCs in critical illnesses such as sepsis.
In various models of vascular injury and organ dysfunction, only a few studies have emerged regarding EPCs in particular as a therapeutic strategy. Transplanted EPCs have been shown to improve survival of mice following liver injury.13 Infusion of EPCs also restored blood flow in a mouse model of hindlimb ischemia.9 A recent prospective randomized trial compared the effects of EPC transplantation in patients with idiopathic pulmonary arterial hypertension versus conventional therapy and showed that after 12 weeks, patients who had received EPCs had a significant improvement in their 6-minute walk test, mean pulmonary artery pressure, pulmonary vascular resistance, and cardiac output.14
Coagulation and Fibrinolysis
A normal physiological function of the endothelium is to provide an antithrombotic surface inhibiting platelet adhesion and clotting, thus facilitating normal blood flow. Under pathophysiological conditions, the endothelium transforms into a prothrombotic surface. A dynamic equilibrium exists between both states that permits a rapid response to an insult and a rapid recovery.15
Anticoagulant Mechanisms
The endothelium has anticoagulant, antiplatelet, and fibrinolytic properties.16 Endothelial cells are the major site for anticoagulant reactions involving thrombin. Thrombin plays a key role in coagulation, including activation of platelets, activation of several coagulation enzymes and cofactors, and stimulation of procoagulation pathways on the endothelial cell surface. In the normal state, there is little thrombin enzyme activity. The surrounding endothelial cell matrix contains heparin sulfate and related glycosaminoglycans that activate antithrombin III. In addition, the subendothelial cell matrix contains dermatan sulfate which promotes the antithrombin activity of heparin cofactor II. Furthermore, microvascular endothelial cells release tissue-factor pathway inhibitor that inhibits the factor VIIa/tissue factor complex, and further contributes to anticoagulation (Figure 101-1).
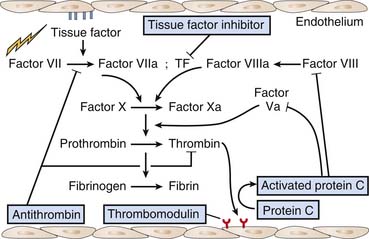
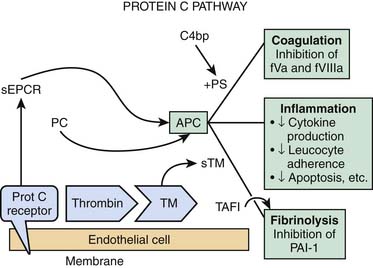
Thrombin activity is also modulated by endothelial cell synthesis of thrombomodulin.17,18 The binding of thrombin to thrombomodulin facilitates the enzyme’s activation of the anticoagulant protein C. Activated protein C (APC) activity is enhanced by cofactor C, also called protein S, which is synthesized by endothelial cells as well as by other cells (see Figure 101-1). APC inhibits factor Va and factor VIIIa. Thrombomodulin (TM) also inhibits prothrombinase activity indirectly by binding factor Xa (Figure 101-2). Protein C has a special receptor on the endothelial cells: endothelial protein C receptor (EPCR). EPCR augments protein C activation approximately twentyfold in vivo by binding protein C and presenting it to the thrombin-TM activation complex. Both EPCR and TM can be found in plasma as soluble proteins. Activated protein C retains its ability to bind EPCR, and this complex appears to be involved in some of the cellular signaling mechanisms that downregulate inflammatory cytokine formation (tumor necrosis factor, interleukin-6). Activated protein C has a variety of antiinflammatory activities. As said, it suppresses inflammatory cytokine during sepsis, inhibits leukocyte adhesion, decreases leukocyte chemotaxis, reduces endothelial cell apoptosis, helps maintain endothelial cell barrier function through activation of the sphingosine-1 phosphate receptor, and minimizes the decrease in blood pressure associated with severe sepsis.19 Another interesting effect of APC is its PAI-1 neutralizing effect. PAI-1 (plasminogen activator inhibitor-1) is a glycoprotein that acts as an acute-phase protein during acute inflammation. Its primary role in vivo is the inhibition of both tissue- and urokinase-type plasminogen activators. PAI-1 is the most efficient inhibitor of activated protein C and thrombin in the absence of heparin. PAI-1 competes with TM for binding with thrombin, which, in combination with its inhibition of activated protein C, makes it a strong local procoagulant by a combined action of displacing and inactivating anticoagulant thrombin from thrombomodulin. In this way PAI-1 has important pathophysiological effects in acute and chronic diseases.20 The thrombin-TM complex also activates thrombin-activatable fibrinolysis inhibitor, a procarboxypeptidase that renders fibrin resistant to clot lysis and neutralizes vasoactive molecules such as complement C5a.21
In addition, platelet adhesion to endothelial cells is markedly inhibited by endothelium-derived prostacyclin.22 The same stimuli that activate platelets, such as thrombin and adenosine diphosphate and adenosine triphosphate (ATP), also act to release prostacyclin from the endothelium, which allows the endothelium to limit the extent of platelet plug formation. The interactions between platelets and endothelium regulate platelet function, coagulation cascades, and local vascular tone.
Microvascular endothelial cells may secrete tissue-type plasminogen activator (t-PA), the powerful thrombolytic agent in frequent clinical use for treatment of coronary thrombotic occlusion.23 t-PA release is stimulated in vivo by norepinephrine, vasopressin, or stasis within the vessel lumen. Thrombin may also stimulate t-PA release, providing a further endothelium-mediated safeguard against uncontrolled coagulation.
Procoagulant Mechanisms
The expression and release of tissue factor is the pivotal step in transforming the endothelium from an anticoagulant to a procoagulant surface.24,25 Tissue factor accelerates factor VIIa-dependent activation of factors X and IX (see Figure 101-1). The synthesis of tissue factor is induced by a number of agonists, including thrombin, endotoxin, several cytokines, shear stress, hypoxia, oxidized lipoproteins, and other endothelial insults. Once endothelial cells expressing tissue factor are exposed to plasma, prothrombinase activity is generated and fibrin is formed on the surface of the cells. Tissue factor can also be found in plasma as a soluble protein. Its role there is not well understood, but it probably plays a role in the initiation of coagulation.
Endothelium-Derived Vasodilators
Nitric Oxide
Furchgott and Zawadzki first postulated the existence of an endothelial derived relaxing factor (EDRF) in 1980, when they noticed that the presence of endothelium was essential for rabbit aortic rings to relax in response to acetylcholine.24 Later, it was determined that the biologic effects of EDRF are mediated by NO.26
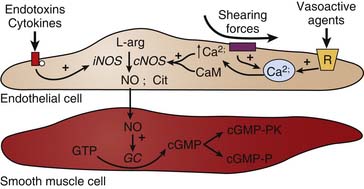
NO is generated from the conversion of L-arginine to NO and L-citrulline by the enzyme nitric oxide synthase (NOS).27 There are two general forms of NOS: constitutive and inducible. In the unstimulated state, NO is continuously produced by constitutive NO synthase (cNOS). The activity of cNOS is modulated by calcium that is released from endoplasmic stores in response to the activation of certain receptors. Substances such as acetylcholine, bradykinin, histamine, insulin, and substance P stimulate NO production through this mechanism. Similarly, shearing forces acting on the endothelium are another important mechanism regulating the release of NO. The inducible form of NOS (iNOS) is not calcium-dependent, but instead is stimulated by the actions of cytokines (e.g., tumor necrosis factor-α [TNFα], interleukins) and/or bacterial endotoxins (e.g., lipopolysaccharide). Induction of iNOS occurs over several hours and results in NO production that may be more than a thousandfold greater than that produced by cNOS. This is an important mechanism in the pathogenesis of inflammation (see Figure 101-2). Inhibition of NOS using competitive analogs of L-arginine drastically reduces endothelium-dependent relaxation in vitro, particularly in large conduit arteries, thereby evoking vasoconstriction. Chronic treatment of animals with NOS inhibitors or suppression of the cNOS gene is reported to induce hypertension.28–30
Once NO is formed by an endothelial cell, it readily diffuses out of the cell and into adjacent smooth muscle cells where it binds and activates the soluble form of guanylyl cyclase, resulting in the production of cyclic guanosine-monophosphate (cGMP) from guanosine-triphosphate.31 cGMP in turn activates a number of cGMP-modulated enzymes (Figure 101-3). Increased cGMP activates a kinase that subsequently leads to the inhibition of calcium influx into the smooth muscle cell, and decreased calcium-calmodulin stimulation of myosin light-chain kinase. This, in turn, decreases the phosphorylation of myosin light chains, thereby decreasing smooth muscle tension development and causing vasodilation. There is also some evidence that increases in cGMP can lead to myosin light chain dephosphorylation by activating the phosphatase. In addition, cGMP-dependent protein kinase phosphorylates K+ channels to induce hyperpolarization and thereby inhibits vasoconstriction.32,33 Interestingly, NO inhibition of platelet aggregation is also related to the increase in cGMP. Drugs that inhibit the breakdown of cGMP such as inhibitors of cGMP-dependent phosphodiesterase (for example, sildenafil) potentiate the effects of NO-mediated actions on the target cell.
< div class='tao-gold-member'>





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


