Endocrine Function
Jeffrey J. Schwartz
Shamsuddin Akhtar
Stanley H. Rosenbaum
Key Points
Related Matter
NIM Thyroid
Vitamin D Metabolism
Catecholamines
Metabolism of Endogenous Catecholamines
Thyroid Gland
The thyroid gland secretes thyroid hormones, thyroxine (T4), and 3,3′,5-triiodothyronine (T3), which are the major regulators of cellular metabolic activity. Thyroid hormones exert a variety of actions by regulating the synthesis and activity of various proteins. They are necessary for proper cardiac, pulmonary, and neurologic function during both health and illness.
Thyroid Metabolism and Function
The production of thyroid hormone is initiated by the active uptake and concentration of iodide in the thyroid gland (Fig. 46-1). Dietary iodine is reduced to iodide in the gastrointestinal (GI) tract. Circulating iodide is taken up by the thyroid gland, where it is then bound to tyrosine residues to form various iodotyrosines. After organification, monoiodotyrosine or diiodotyrosine is coupled enzymatically by thyroid peroxidase to form either T3 or T4. These hormones are attached to the thyroglobulin protein and stored as colloid in the gland. The release of T3 and T4 from the gland is accomplished through proteolysis from the thyroglobulin and diffusion into the circulation. Thyrotropin (thyroid-stimulating hormone [TSH]) is produced in the anterior pituitary gland, and its secretion is regulated by thyrotropin-releasing hormone produced in the hypothalamus. TSH is responsible for maintaining the uptake of iodide and proteolytic release of thyroid hormone. Excess iodide inhibits the synthesis and secretion of thyroid hormone. Circulating thyroid hormone inhibits thyroid-releasing hormone and TSH secretion in a negative-feedback loop. The thyroid gland is solely responsible for the daily secretion of T4 (80 to 100 μg/day). The half-life of T4 in the circulation is 6 to 7 days.
Approximately 80% of T3 is produced by the extrathyroidal deiodination of T4 and 20% is produced by direct thyroid secretion. The half-life of T3 is 24 to 30 hours. Most of the effects of thyroid hormones are mediated by the more potent and less protein-bound T3. The degree to which these hormones are protein bound in the circulation is the major factor influencing their activity and degradation. T4 is metabolized by monodeiodination to either T3 or reverse T3 (rT3). T3 is biologically active, whereas rT3 is inactive. The major fraction of circulating hormone is bound to thyroxine-binding globulin (TBG), with a smaller fraction bound to albumin and transthyretin. Less than 0.1% is present as free, unbound hormone. Changes in serum-binding protein concentrations have a major effect on total T3 and T4 serum concentrations. The plasma normally contains 5 to 12 μg/dL of T4 and 60 to 180 ng/dL of T3. Many drugs can affect thyroid function, including amiodarone and dopamine.1
Although the thyroid hormone is important to many aspects of growth and function, the anesthesiologist is most often concerned with the cardiovascular manifestations of thyroid disease.2 Thyroid hormones affect tissue responses to sympathetic stimuli and increase the intrinsic contractile state of cardiac muscle.
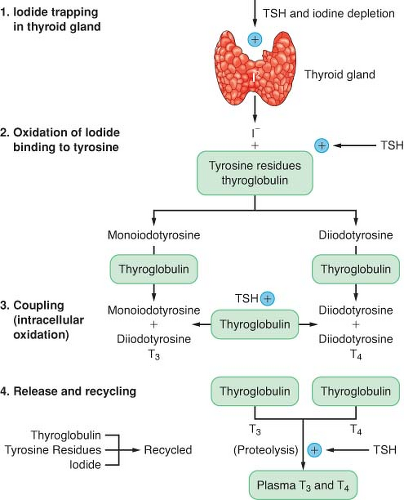 Figure 46-1. Thyroid hormone biosynthesis consists of four stages: (1) organification, (2) binding, (3) coupling, and (4) release. TSH, thyroid-stimulating hormone; T3, triiodothyronine; T4, thyroxine. |
Table 46-1. Tests of Thyroid Gland Function | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
β-adrenergic receptors are increased in number, and cardiac α-adrenergic receptors are decreased by thyroid hormone.3
Tests of Thyroid Function
Serum Thyroxine
The serum T4 assay is a standard test for evaluation of thyroid gland function (Table 46-1). The total T4 is elevated in approximately 90% of patients with hyperthyroidism, and it is low in 85% of those who are hypothyroid. The concentration of T4 is measured by radioimmunoassay (RIA). The serum T4 concentration is influenced by thyroid hormone protein–binding capacity. An increase or decrease in TBG levels or in protein binding may therefore alter the total T4 but not the concentration of the free T4. Because of the effect of TBG on circulating total T4, the T4 levels should never be used alone to evaluate thyroid disease. Elevations in the TBG concentration are the most common cause of hyperthyroxinemia in euthyroid patients. Increases in TBG due to acute liver disease, pregnancy, or drugs (oral contraceptives, exogenous estrogens, clofibrate, opioids) may be the causative factor. Because a total T4 can be misleadingly high in euthyroidism or normal in hypothyroidism, some measure of free thyroid hormone activity (free T4) must also be used.
Serum Triiodothyronine
The serum T3 is also measured by RIA. Serum T3 levels are often determined to detect disease in patients with clinical evidence of hyperthyroidism in the absence of elevations of T4. T3 may be the only thyroid hormone produced in excess. T3 concentrations may be depressed by factors that impair the peripheral conversion of T4 to T3 (sick euthyroid syndrome). In 50% of hypothyroid patients, the serum T3 concentration is low; in the remaining 50%, it is normal.
Tests for Assessing Thyroid Hormone Binding
Because conventional assays measure total hormone levels, which can be affected by protein binding without affecting free hormone levels, it is necessary to find some measure of thyroid-binding proteins to correctly interpret total thyroxine levels. A direct measurement of unbound T3 and T4 can be performed by equilibrium dialysis or by unbound immunoassays. Indirect measurement of free hormone levels can be calculated by multiplying the total hormone level by the thyroid hormone binding ratio, which can be calculated from the T3-resin uptake. This product is the free T3 or T4 index. The T3 uptake test measures the ability of the patient’s serum to bind exogenously introduced T3 and reflects the amount of TBG and the extent of T3 saturation on TBG. The T3 uptake is inversely related to the degree of unsaturation of TBG.
Thyroid-Stimulating Hormone
The radioimmunoassay for this hormone is sensitive and specific enough to become the first test in evaluating suspected thyroid dysfunction. It is often higher than 20 μIU/mL in primary hypothyroidism (normal 0.4 to 4.5 μIU/mL). Hyperthyroidism can be suspected from depressed TSH levels. A condition characterized by elevated TSH and normal T4 may represent subclinical hypothyroidism. A low TSH level in a clinically hypothyroid patient indicates disease at the pituitary or hypothalamic level. The goal of thyroid replacement therapy is to normalize TSH levels.3 Starvation, fever, stress, corticosteroids, and T3 or T4 can all depress TSH levels.
Radioactive Iodine Uptake
The thyroid gland has the ability to concentrate large amounts of inorganic iodide. The oral administration of radioactive iodine (131I) can be used to indicate thyroid gland activity. Thyroid uptake is elevated in hyperthyroidism unless the hyperthyroidism is caused by thyroiditis, in which case the uptake is low or absent. Because of overlap in values, it is difficult to distinguish euthyroid from hypothyroid people. Radioactive iodide uptake may be increased by a variety of factors, including dietary iodine deficiency, renal failure, and congestive heart failure. Because uptake is under TSH control, elevated free T4 levels and corticosteroids decrease radioactive iodide uptake. Functioning (“hot”) thyroid tissue is rarely malignant. Nonfunctioning (“cold”) tissue may be malignant or benign.
Hyperthyroidism
Hyperthyroidism results from the exposure of tissues to excessive amounts of thyroid hormone (Table 46-2). The most common cause is the multinodular diffuse goiter of Graves disease. This typically occurs between the ages of 20 and 40 years and is predominant in women. Most patients with this condition demonstrate a syndrome characterized by diffuse glandular enlargement, ophthalmopathy, dermopathy, and clubbing of the fingers. A thyroid-stimulating autoantibody may be present. Thyroid adenoma is the second most common cause. Another cause of increased thyroid hormone synthesis is thyroiditis. Subacute thyroiditis frequently
follows a respiratory illness and is characterized by a viral-like illness with a firm, painful gland. This type of thyroiditis is frequently treated with anti-inflammatory agents alone. Rarely, subacute thyroiditis may occur in a patient with a normal-sized painless gland. Hashimoto thyroiditis is a chronic autoimmune disease that usually produces hypothyroidism but may occasionally produce hyperthyroidism. Hyperthyroidism may also be associated with pregnancy, 131I therapy, thyroid carcinoma, trophoblastic tumors, or TSH-secreting pituitary adenomas. Iatrogenic hyperthyroidism may follow thyroid hormone replacement or may occur after iodide exposure (angiographic contrast media) in patients with chronically low iodide intake (Jod-Basedow phenomenon). The antiarrhythmic agent amiodarone is iodine rich and is another cause of iodine-induced thyrotoxicosis.4
follows a respiratory illness and is characterized by a viral-like illness with a firm, painful gland. This type of thyroiditis is frequently treated with anti-inflammatory agents alone. Rarely, subacute thyroiditis may occur in a patient with a normal-sized painless gland. Hashimoto thyroiditis is a chronic autoimmune disease that usually produces hypothyroidism but may occasionally produce hyperthyroidism. Hyperthyroidism may also be associated with pregnancy, 131I therapy, thyroid carcinoma, trophoblastic tumors, or TSH-secreting pituitary adenomas. Iatrogenic hyperthyroidism may follow thyroid hormone replacement or may occur after iodide exposure (angiographic contrast media) in patients with chronically low iodide intake (Jod-Basedow phenomenon). The antiarrhythmic agent amiodarone is iodine rich and is another cause of iodine-induced thyrotoxicosis.4
Table 46-2. Causes of Hyperthyroidism | ||
|---|---|---|
|
The major manifestations of hyperthyroidism are weight loss, diarrhea, skeletal muscle weakness and stiffness, warm and moist skin, heat intolerance, and nervousness. Cardiovascular manifestations include increased left ventricular contractility and ejection fraction, tachycardia, elevated systolic blood pressure, and decreased diastolic blood pressure. Hypercalcemia, thrombocytopenia, and a mild anemia may be present. Elderly patients may present with heart failure, atrial fibrillation, or other cardiac dysrhythmias. They may also present with apathetic hyperthyroidism characterized by depression and withdrawal, without the usual systemic signs or symptoms.
Treatment and Anesthetic Considerations
The most important goal in managing the hyperthyroid patient is to make the patient euthyroid before any surgery, if possible. The drugs propylthiouracil and methimazole are thiourea derivatives that inhibit organification of iodide and the synthesis of thyroid hormone.5 Propylthiouracil also decreases the peripheral conversion of T4 to T3. Normal thyroid glands usually contain a store of hormone that is large enough to maintain a euthyroid state for several months, even if synthesis is abolished. Therefore, hyperthyroid patients are unlikely to be regulated to a euthyroid state with antithyroid drugs alone in <6 to 8 weeks. Toxic reactions from these drugs are uncommon but include skin rash, nausea, fever, agranulocytosis, hepatitis, and arthralgias.
Inorganic iodide inhibits iodide organification and thyroid hormone release—the Wolff-Chaikoff effect. Iodide is also effective in reducing the size of the hyperplastic gland and has a role in the preparation of the patient for emergency thyroid surgery. Antithyroid drugs should be started before iodide treatment because of the possibility of worsening the thyrotoxicosis.
β-adrenergic antagonists are effective in attenuating the manifestations of excessive sympathetic activity and should be used in all hyperthyroid patients unless contraindicated. β-adrenergic blockade alone does not inhibit hormone synthesis, but specifically propranolol does impair the peripheral conversion of T4 to T3 over 1 to 2 weeks. Propranolol given over 12 to 24 hours decreases tachycardia, heat intolerance, anxiety, and tremor. Any β-blocker may be used, and long-acting agents may be more convenient. The combination of propranolol (in doses titrated to effect) plus potassium iodide (2 to 5 drops every 8 hours) is frequently used before surgery to ameliorate cardiovascular symptoms and reduce circulating concentrations of T4 and T3. Preoperative preparation usually requires 7 to 14 days.
Heart failure secondary to poorly controlled paroxysmal atrial fibrillation may improve with slowing of the ventricular rate, but abnormalities of left ventricular function secondary to hyperthyroidism may not be corrected with the use of β-antagonists. If a hyperthyroid patient with clinically apparent disease requires emergency surgery, β-adrenergic blockade should be administered to achieve a heart rate <90 beats per minute. β-blockers do not prevent thyroid storm. Glucocorticoids such as dexamethasone (8 to 12 mg/day) are used in the management of severe thyrotoxicosis because they reduce thyroid hormone secretion and the peripheral conversion of T4 to T3.
Iopanoic acid, a radiographic contrast agent that decreases peripheral conversion of T4 and releases iodine that inhibits synthesis, is useful for emergency preparation.
Radioactive iodine therapy is an effective treatment for some patients with thyrotoxicosis.6 However, it should not be administered to patients who are pregnant because it crosses the placenta and may destroy the fetal thyroid. A side effect of radioiodine therapy is hypothyroidism; 10% to 60% of cases occur in the first year of therapy and an additional 2% occur per year thereafter.
A variety of anesthetic techniques and drugs have been used for hyperthyroid patients undergoing surgery. All antithyroid medications are continued through the morning of surgery. The goal of intraoperative management in the hyperthyroid patient is to achieve a depth of anesthesia that prevents an exaggerated sympathetic response to surgical stimulation while avoiding the administration of medication that stimulates the sympathetic nervous system. Pancuronium should be avoided. It is best to avoid using ketamine, even when a patient is clinically euthyroid. Hypotension that occurs during surgery is best treated with direct-acting vasopressors rather than a medication that provokes the release of catecholamines. The incidence of myasthenia gravis is increased in hyperthyroid patients; thus, the initial dose of muscle relaxant should be reduced and a twitch monitor should be used to titrate subsequent doses. Regional anesthesia is an excellent alternative when appropriate; however, epinephrine-containing solutions should be avoided.
Thyroid storm is a life-threatening exacerbation of hyperthyroidism that most commonly develops in the undiagnosed or untreated hyperthyroid patient because of the stress of surgery or nonthyroid illness.7 Operating on an acutely hyperthyroid gland may provoke thyroid storm, although this is probably not due to mechanical release of hormone.8 Its manifestations include hyperthermia, tachycardia, dysrhythmias, myocardial ischemia, congestive heart failure, agitation, and confusion. It must be distinguished from, or considered with, pheochromocytoma,
malignant hyperthermia, and light anesthesia. Although free T4 levels are often markedly elevated, no laboratory test is diagnostic. Treatment involves large doses of propylthiouracil and supportive measures to control fever and restore intravascular volume. Hemodynamic monitoring (pulmonary artery catheter, arterial catheter) is especially useful in guiding the treatment of patients with significant left ventricular dysfunction (Table 46-3). Again, it is essential to remove or treat the precipitating event.
malignant hyperthermia, and light anesthesia. Although free T4 levels are often markedly elevated, no laboratory test is diagnostic. Treatment involves large doses of propylthiouracil and supportive measures to control fever and restore intravascular volume. Hemodynamic monitoring (pulmonary artery catheter, arterial catheter) is especially useful in guiding the treatment of patients with significant left ventricular dysfunction (Table 46-3). Again, it is essential to remove or treat the precipitating event.
Table 46-3. Management of Thyroid Storm | ||
|---|---|---|
|
Anesthesia for Thyroid Surgery
Hypothyroidism
Hypothyroidism is a relatively common disease (0.3% to 5% of the adult population) that results from inadequate circulating levels of T4 or T3 or both.13 The development of hypothyroidism is often slow and progressive, making the clinical diagnosis difficult, especially in more subtle cases. Hypofunctioning of the thyroid gland has many causes (Table 46-4). Primary failure of the thyroid gland refers to decreased production of thyroid hormone, despite adequate TSH production, and accounts for 95% of all cases of thyroid dysfunction. The remainder of the cases are caused by either hypothalamic or pituitary disease (secondary hypothyroidism) and are associated with other pituitary deficiencies.
A lack of thyroid hormone produces a variety of signs and symptoms. These early findings are often nonspecific and difficult to recognize. A history of radioiodine therapy, external neck irradiation, or the presence of a goiter is helpful in diagnosis. There is a generalized reduction in metabolic activity, resulting in lethargy, slow mental functioning, cold intolerance, and slow movements. The cardiovascular manifestations of hypothyroidism reflect the importance of thyroid hormone for myocardial contractility and catecholamine function. These patients exhibit bradycardia, decreased cardiac output, and increased peripheral resistance.14 The accumulation of a cholesterol-rich pericardial fluid produces
low voltage on the electrocardiogram (ECG). Heart failure only rarely occurs in the absence of coexisting heart disease. Angina pectoris itself is unusual in hypothyroidism but can appear when thyroid hormone treatment is initiated. Ventilatory responsiveness to hypoxia and hypercapnia is depressed in hypothyroid patients. This depression is potentiated by sedatives, opioids, and general anesthesia. Postoperative ventilatory failure requiring prolonged ventilation is rarely seen in hypothyroid patients in the absence of coexisting lung disease, obesity, or myxedema coma. Other abnormalities found in hypothyroidism include anemia, coagulopathy, hypothermia, sleep apnea, and impaired renal free water clearance with hyponatremia. Decreased GI motility can compound the effect of postoperative ileus. In long-standing or severe disease, the stress response may be blunted and adrenal depression may occur.
low voltage on the electrocardiogram (ECG). Heart failure only rarely occurs in the absence of coexisting heart disease. Angina pectoris itself is unusual in hypothyroidism but can appear when thyroid hormone treatment is initiated. Ventilatory responsiveness to hypoxia and hypercapnia is depressed in hypothyroid patients. This depression is potentiated by sedatives, opioids, and general anesthesia. Postoperative ventilatory failure requiring prolonged ventilation is rarely seen in hypothyroid patients in the absence of coexisting lung disease, obesity, or myxedema coma. Other abnormalities found in hypothyroidism include anemia, coagulopathy, hypothermia, sleep apnea, and impaired renal free water clearance with hyponatremia. Decreased GI motility can compound the effect of postoperative ileus. In long-standing or severe disease, the stress response may be blunted and adrenal depression may occur.
Table 46-4. Causes of Hypothyroidism | ||
|---|---|---|
|
Treatment and Anesthetic Considerations
Treatment of symptomatic hypothyroidism is with hormone replacement therapy.15 Controversy remains regarding the preoperative anesthetic management of the hypothyroid patient. Although it seems logical, given the multisystem effects of thyroid hormone, to recommend that all hypothyroid surgical candidates be restored to a euthyroid state before surgery, such a recommendation is, in general, based on individual case reports. There have been few controlled studies to support the position that most hypothyroid patients are unusually sensitive to anesthetic drugs, have prolonged recovery times, or have a higher incidence of cardiovascular instability or collapse.
No increase in serious complications in patients with mild or moderate hypothyroidism undergoing general anesthesia has been noted.16 One study noted a higher incidence of intraoperative hypotension and postoperative GI and neuropsychiatric complications in mild and moderately hypothyroid patients undergoing noncardiac surgery, but still noted there were no compelling clinical reasons to postpone surgery in these patients.17 Surgery in severely hypothyroid patients should be postponed when possible until these patients are at least partially treated.
The management of hypothyroid patients with symptomatic coronary artery disease has been a subject of particular controversy.18 The need for thyroid hormone replacement therapy must be weighed against the risk of precipitating myocardial ischemia. Several studies and a literature review found no differences in the frequency of intraoperative or postoperative complications when mild or moderately hypothyroid patients underwent cardiac surgery. In symptomatic patients or unstable patients with cardiac ischemia, thyroid replacement should probably be delayed until after coronary revascularization.
A number of anesthetic medications have been used without difficulty in hypothyroid patients. Although ketamine has been proposed as the ideal induction agents, all intravenous induction agents have been used in the hypothyroid patient. The maintenance of anesthesia may be safely achieved with either intravenous or inhaled anesthetics. There appears to be little, if any, decrease in the minimum alveolar concentration for volatile agents. Regional anesthesia is a good choice in the hypothyroid patient, provided the intravascular volume is well maintained. Monitoring is directed toward the early recognition of hypotension, congestive heart failure, and hypothermia. Scrupulous attention should be paid to maintaining normal body temperature.
Table 46-5. Management of Myxedema | ||
|---|---|---|
|
Myxedema coma represents a severe form of hypothyroidism characterized by stupor or coma, hypoventilation, hypothermia, hypotension, and hyponatremia. This is a medical emergency with a high mortality rate (25% to 50%) and, as such, requires aggressive therapy (Table 46-5). Only lifesaving surgery should proceed in the face of myxedema coma. Intravenous thyroid replacement is initiated as soon as the clinical diagnosis is made. An intravenous loading dose of T4 (sodium levothyroxine, 200 to 300 μg) is given initially and followed by a maintenance dose of T4, 50 to 200 μg/day intravenously.19 Alternatively, T3 may be used because it has a more rapid onset. Improvements in heart rate, blood pressure, and body temperature may occur within 24 hours. However, replacement therapy with either form of thyroid hormone may precipitate myocardial ischemia. There is also an increased likelihood of acute primary adrenal insufficiency in these patients, and they should receive stress doses of hydrocortisone. Steroid replacement continues until normal adrenal function can be confirmed.
Parathyroid Glands
Calcium Physiology
The normal adult body contains approximately 1 to 2 kg of calcium (Ca2+), of which 99% is in the skeleton.20 Plasma calcium is present in three forms: (a) a protein-bound fraction (50%), (b) an ionized fraction (45%), and (c) a diffusible but nonionized fraction (5%) that is complexed with phosphate, bicarbonate, and citrate (see Chapter 14). This division is interesting because it is the ionized fraction that is physiologically active and homeostatically regulated. The normal total serum calcium concentration is 8.8 to 10.4 mg/dL. Albumin binds approximately 90% of the protein-bound fraction of calcium, and total serum Ca2+ consequently depends on albumin levels. In general, an increase or decrease in albumin of 1 g/dL is associated with a parallel change in total serum Ca2+ of 0.8 mg/dL. The serum ionized Ca2+ concentration is affected by temperature and blood pH through alterations in Ca2+ protein binding to albumin. Acidosis decreases protein binding (increases ionized Ca2+), and alkalosis increases protein binding (decreases ionized Ca2+). The concentration of free Ca2+ ion is of critical importance in regulating skeletal muscle contraction, coagulation, neurotransmitter release, endocrine secretion, and a variety of other cellular functions. As a consequence, the maintenance of serum Ca2+ concentration is subject
to tight hormonal control by parathyroid hormone (PTH) and vitamin D (Fig. 46-2).
to tight hormonal control by parathyroid hormone (PTH) and vitamin D (Fig. 46-2).
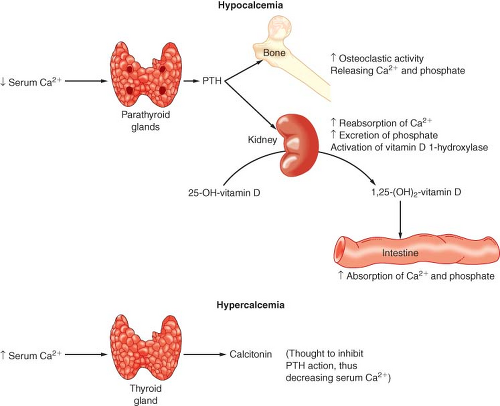 Figure 46-2. Parathyroid hormone (PTH) and vitamin D metabolism and action. 25-OH, 25-hydroxycholecalciferol; 1,25-(OH)2, 1,25-dihydroxycholecalciferol. (From McClatchey KD. Clinical laboratory medicine. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2002.) |
PTH secretion is primarily regulated by the serum ionized Ca2+ concentration. This negative-feedback mechanism is exquisitely sensitive in maintaining calcium levels in a normal range. Release of PTH is also influenced by phosphate, magnesium, and catecholamine levels. Acute hypomagnesemia directly stimulates PTH release, whereas chronic magnesium depletion appears to inhibit proper functioning of the parathyroid gland. The plasma phosphate concentration has an indirect influence on PTH secretion by causing reciprocal changes in the serum ionized Ca2+ concentration.
Vitamin D is absorbed from the GI tract and can be produced enzymatically by ultraviolet irradiation of the skin. Vitamin D (cholecalciferol) is made from cholesterol metabolites and is inactive. Calciferol is hydroxylated in the liver to 25-hydroxycholecalciferol (25-OHD) and in the kidney is further hydroxylated to 1,25-dihydroxycholecalciferol [1,25(OH)2D] or 24,25-dihydroxycholecalciferol [24,25(OH)2D]. 25-OHD is the major circulating form of vitamin D. The synthesis of this hormone is not regulated by a hormone or by Ca2+ or phosphate levels. 1,25(OH)2D and 24,25(OH)2D are the major active metabolites of vitamin D, and their production is reciprocally regulated at the kidney. Hypocalcemia and hypophosphatemia cause an increased production of 1,25(OH)2D and a decreased production of 24,25(OH)2D. 1,25(OH)2D stimulates bone, kidney, and intestinal absorption of calcium and phosphate. Vitamin D deficiency can lead to decreased intestinal absorption of Ca2+ and secondary hyperparathyroidism.
Hyperparathyroidism
Primary hyperparathyroidism is most commonly due to a benign parathyroid adenoma (90% of cases) or hyperplasia (9%) and very rarely to a parathyroid carcinoma.21 Primary hyperparathyroidism may also exist as part of a multiple endocrine neoplastic (MEN) syndrome. Hyperplasia usually involves all four glands. Although most patients with primary hyperparathyroidism are
hypercalcemic, most are asymptomatic at the time of diagnosis. When symptoms occur, they usually result from the hypercalcemia that accompanies the disease. Primary hyperparathyroidism occurring during pregnancy is associated with a high maternal and fetal morbidity rate (50%). The placenta allows the fetus to concentrate calcium, promoting fetal hypercalcemia and leading to hypoparathyroidism in the newborn. Pregnant women with primary hyperparathyroidism should generally be treated with surgery.
hypercalcemic, most are asymptomatic at the time of diagnosis. When symptoms occur, they usually result from the hypercalcemia that accompanies the disease. Primary hyperparathyroidism occurring during pregnancy is associated with a high maternal and fetal morbidity rate (50%). The placenta allows the fetus to concentrate calcium, promoting fetal hypercalcemia and leading to hypoparathyroidism in the newborn. Pregnant women with primary hyperparathyroidism should generally be treated with surgery.
Hypercalcemia is responsible for a broad spectrum of signs and symptoms. Nephrolithiasis is the most common manifestation, occurring in 60% to 70% of patients. Polyuria and polydipsia are also common complaints. An increase in bone turnover may lead to generalized demineralization and subperiosteal bone resorption; however, only a small group of patients (10% to 15%) have clinically significant bone disease. Patients may experience generalized skeletal muscle weakness and fatigability, epigastric discomfort, peptic ulceration, and constipation. Psychiatric manifestations include depression, memory loss, confusion, or psychosis. Between 20% and 50% of patients are hypertensive, but this usually resolves with successful treatment of the disease. Cardiac function is enhanced in the early stages of hypercalcemia. Calcium flux into the cells is reflected in the plateau phase of the action potential (phase 2). As extracellular calcium increases, the inward flux is more rapid, and phase 2 is shortened (see Chapter 10). The corresponding ECG change is a shorter QT interval. Cardiac contractility may increase until a level between 15 and 20 mg/dL is reached. At this point, there is a prolongation of the PR segment and QRS complex that can result in heart block or bundle-branch block. Bradycardia also occurs.
An elevated serum Ca2+ concentration is a valuable diagnostic indicator of primary hyperparathyroidism. The serum phosphate concentration is nonspecific, with many patients having normal or near-normal levels. The reported incidence of hyperchloremic acidosis varies widely in primary hyperparathyroidism, but most patients usually have a serum chloride concentration in excess of 102 mEq/L. Rarely does a patient with hypercalcemia secondary to ectopic PTH production (malignancy) present with hyperchloremic acidosis. The definitive diagnosis of primary hyperparathyroidism is made by RIA demonstration of an elevation in PTH levels in the presence of hypercalcemia. An elevated nephrogenous cyclic adenosine monophosphate is noted in >90% of patients with primary hyperparathyroidism.
Hypercalcemia may also result from the ectopic production of PTH or PTH-like substances from lung, genitourinary, breast, GI, and lymphoproliferative malignancies. Tumors may also produce hypercalcemia through direct bone resorption or the production of osteoclast-activating factor. In the absence of a clinically obvious neoplasm, there may be difficulty in differentiating between PTH-producing malignancies and primary hyperparathyroidism. PTH fragments from malignant tissue differ from native PTH, so precise clinical identification may aid in distinguishing between ectopic PTH production and primary hyperparathyroidism.
Secondary hyperparathyroidism represents an increase in parathyroid function as a result of conditions that produce hypocalcemia or hyperphosphatemia. Chronic renal disease is a common cause of hyperphosphatemia (due to decreased phosphate excretion) and decreased vitamin D metabolism. The hypocalcemia that results leads to an increased production of PTH. GI disorders accompanied by malabsorption may also lead to a secondary increase in parathyroid activity. Tertiary hyperparathyroidism refers to the development of hypercalcemia in a patient who has had prolonged secondary hyperparathyroidism that has caused adenomatous changes in the parathyroid gland and unregulated PTH.
Treatment and Anesthetic Considerations
Surgery is the treatment of choice for the patient with symptomatic disease. However, there is considerable controversy surrounding the choice of treatment in the asymptomatic patient. It is not clear whether mild primary hyperparathyroidism decreases longevity. Surgery is often chosen over medical therapy because it offers definitive treatment and is generally safe.
Preoperative preparation focuses on the correction of intravascular volume and electrolyte irregularities. It is particularly important to evaluate the patient with chronic hypercalcemia for abnormalities of the renal, cardiac, or central nervous systems. Emergency treatment of hypercalcemia is undertaken before surgery when the serum Ca2+ concentration exceeds 15 mg/dL (7.5 mEq/L). Lowering of the serum Ca2+ concentration is initially accomplished by expanding the intravascular volume and establishing a sodium diuresis. This is achieved with the intravenous administration of normal saline and furosemide. Rehydration alone is capable of lowering the serum Ca2+ level by ≥2 mg/dL. Hydration dilutes the serum Ca2+, and sodium diuresis promotes Ca2+ excretion through an inhibition of sodium and Ca2+ resorption in the proximal tubule. Hypokalemia and hypomagnesemia may result.
Another element in the treatment of hypercalcemia is the correction of hypophosphatemia. Hypophosphatemia increases GI absorption of Ca2+, stimulates the breakdown of bone, and impairs the uptake of Ca2+ by bone. Low serum phosphate levels impair cardiac contractility and may contribute to congestive heart failure. Hypophosphatemia also causes skeletal muscle weakness, hemolysis, and platelet dysfunction.
Other medications that have a role in lowering the serum Ca2+ include bisphosphonates, mithramycin, calcitonin, and glucocorticoids. Bisphosphonates are pyrophosphate analogs that inhibit osteoclast action. They are the drugs of choice for severe hypercalcemia. Toxic effects include fever and hypophosphatemia. Mithramycin, a cytotoxic agent, inhibits PTH-induced osteoclast activity and can lower the serum Ca2+ levels by ≥2 mg/dL in 24 to 48 hours. Toxic effects include azotemia, hepatotoxicity, and thrombocytopenia. Calcitonin is useful in transiently lowering the serum Ca2+ level 2 to 4 mg/dL through direct inhibition of osteoclastic bone resorption. The advantages of calcitonin are the mild side effects (urticaria, nausea) and the rapid onset of activity. Calcitonin resistance usually develops within 24 to 48 hours. Glucocorticoids are effective in lowering the serum Ca2+ concentration in several conditions (sarcoidosis, some malignancies, hyperthyroidism, vitamin D intoxication) through their actions on osteoclast bone resorption, GI absorption of calcium, and the urinary excretion of calcium. Glucocorticoids are usually of no benefit in the treatment of primary hypercalcemia. Finally, hemodialysis or peritoneal dialysis can be used to lower the serum Ca2+ level when alternative regimens are ineffective or contraindicated.
There is no evidence that a specific anesthetic drug or technique has advantages over another. A thorough knowledge of the clinical manifestations attributable to hypercalcemia is of the greatest value in choosing an anesthetic technique. Special monitoring is usually not required. Because of the unpredictable response to neuromuscular blocking drugs in the hypercalcemic patient, a conservative approach to muscle paralysis makes sense. There is an increased requirement for vecuronium, and probably all nondepolarizing muscle relaxants, during onset of
neuromuscular blockade.22 Careful positioning of the osteopenic patient is necessary to avoid pathologic bone fractures.
neuromuscular blockade.22 Careful positioning of the osteopenic patient is necessary to avoid pathologic bone fractures.
Anesthesia for Parathyroid Surgery
General anesthesia is most commonly used for parathyroid surgery. Minimally invasive parathyroidectomy is superior to conventional bilateral cervical exploration in patients with sporadic primary hyperparathyroidism23 and can usually be performed under bilateral cervical plexus block.24 Some centers use an intraoperative rapid PTH assay to help determine when a hyperfunctioning gland has been removed. There is in vitro, but no clinical,25 evidence that propofol can interfere with the assay, so many surgeons prefer that propofol not be used within 15 minutes of an assay. Postoperative complications include RLN injury, bleeding, and transient or complete hypoparathyroidism. Unilateral RLN is characterized by hoarseness and usually requires no intervention. Bilateral RLN injury is a rare complication, producing aphonia and requiring immediate tracheal intubation.
After successful parathyroidectomy, a decrease in the serum Ca2+ level should be observed within 24 hours. Patients with significant preoperative bone disease may have hypocalcemia after removal of the PTH-secreting glands. This “hungry bone” syndrome comes as a result of the rapid remineralization of bone. Thus, serum Ca2+, magnesium, and phosphorus levels should be closely monitored until stable. The serum Ca2+ nadir usually occurs within 3 to 7 days.
Hypoparathyroidism
An underproduction of PTH or resistance of the end-organ tissues to PTH results in hypocalcemia (<8 mg/dL).26 The normal physiologic response to hypocalcemia is an increase in PTH secretion and 1,25(OH)2D synthesis, with an increase in Ca2+ mobilization from bone, GI absorption, and renal tubule reclamation. The most common cause of acquired PTH deficiency is inadvertent removal of the parathyroid glands during thyroid or parathyroid surgery. Other causes of acquired hypoparathyroidism include 131I therapy for thyroid disease, neck trauma, granulomatous disease, or an infiltrating process (malignancy or amyloidosis). Severe hypomagnesemia (<0.8 mEq/L) from any cause can produce hypocalcemia by suppressing PTH secretion and interfering with PTH action. Renal insufficiency leads to phosphorus retention and impaired 1,25(OH)2D synthesis, which results in hypocalcemia. These patients are commonly treated with vitamin D, which increases intestinal calcium absorption and suppresses secondary increases in PTH secretion. Hypocalcemia due to pancreatitis and burns results from the suppression of PTH and from the sequestration of calcium.
Clinical Features and Treatment
The clinical features of hypoparathyroidism are a manifestation of hypocalcemia. Neuronal irritability and skeletal muscle spasms, tetany, or seizures reflect a reduced threshold of excitation. Latent tetany may be demonstrated by eliciting the Chvostek or Trousseau sign. Chvostek sign is a contracture of the facial muscle produced by tapping the facial nerve as it passes through the parotid gland. Trousseau sign is contraction of the fingers and wrist after application of a blood pressure cuff inflated above the systolic blood pressure for approximately 3 minutes. Other common complaints of hypocalcemia include fatigue, depression, paresthesias, and skeletal muscle cramps. The acute onset of hypocalcemia after thyroid or parathyroid surgery may manifest as stridor and apnea. Cardiovascular manifestations of hypocalcemia include congestive heart failure, hypotension, and a relative insensitivity to the effects of β-adrenergic agonists (see Chapter 10). Delayed ventricular repolarization results in a prolonged QT interval on the ECG. Although prolongation of the QT interval may be a reliable sign of hypocalcemia in an individual patient, the ECG is relatively insensitive for the detection of hypocalcemia.
The treatment of hypoparathyroidism consists of electrolyte replacement. The objective is to have the patient’s clinical symptoms under control before anesthesia and surgery. Hypocalcemia caused by magnesium depletion is treated by correcting the magnesium deficit. Serum phosphate excess is corrected by the removal of phosphate from the diet and the oral administration of phosphate-binding resins (aluminum hydroxide). The urinary excretion of phosphate can be increased with a saline volume infusion. Ca2+ deficiencies are corrected with Ca2+ supplements or vitamin D analogs. Patients with severe symptomatic hypocalcemia are treated with intravenous calcium gluconate (10 to 20 mL of 10% solution) given over several minutes and followed by a continuous infusion (1 to 2 mg/kg/hour) of elemental Ca2+. The correction of serum Ca2+ levels should be monitored by measuring serum Ca2+ concentrations and following clinical symptoms. When oral or intravenous calcium is inadequate to maintain a normal serum–ionized calcium level, vitamin D is added to the regimen.
Adrenal Cortex
The adrenal cortex functions to synthesize and secrete three types of hormones. Endogenous and dietary cholesterol is used in the adrenal biosynthesis of glucocorticoids (cortisol), mineralocorticoids (aldosterone and 11-deoxycorticosterone), and androgens (dehydroepiandrosterone). Cortisol and aldosterone are the two essential hormones, whereas adrenal androgens are of relatively minor physiologic significance in adults. The major biologic effects of adrenal cortical hyperfunction or hypofunction occur as a result of cortisol or aldosterone excess or deficiency. Abnormal function of the adrenal cortex may render a patient unable to respond appropriately during a period of surgical stress or critical illness.
Glucocorticoid Physiology
Cortisol (hydrocortisone) is the most potent endogenous glucocorticoid and is produced by the inner portions of the adrenal cortex. Cortisone is a glucocorticoid produced in small amounts. Cortisol is produced under the control of adrenocorticotropic hormone (ACTH; corticotropin), a polypeptide synthesized and released by the anterior pituitary gland. Glucocorticoids exert their biologic effects by diffusing into the cytoplasm of target cells and combining with specific high-affinity receptor proteins.
The daily production of endogenous cortisol is approximately 20 mg. The maximal output is 150 to 300 mg. Most of the circulating hormone is bound to the α-globulin cortisol-binding globulin. It is the relatively small amount of free hormone that exerts the biologic effects. Endogenous glucocorticoids are inactivated primarily by the liver and are excreted in the urine as 17-hydroxycorticosteroids. Cortisol is also filtered at the glomerulus and may be excreted unchanged in the urine. Although the rate of cortisol secretion is decreased by approximately 30% in the elderly patient, plasma cortisol levels remain in a normal range because of a corresponding decrease in hepatic and renal clearance.
Cortisol secretion is directly controlled by ACTH, which in turn is regulated by the corticotropin-releasing factor from the hypothalamus. ACTH is synthesized in the pituitary gland from a precursor molecule that also produces β-lipotropin and β-endorphin. The secretion of ACTH and corticotropin-releasing factor is governed chiefly by glucocorticoids, the sleep–wake cycle, and stress. Cortisol is the most potent regulator of ACTH secretion, acting by a negative-feedback mechanism to maintain cortisol levels in a physiologic range. ACTH release follows a diurnal pattern, with maximal activity occurring soon after awakening. This diurnal pattern of activity occurs in normal subjects and in those with adrenal insufficiency. Psychological or physical stress (trauma, surgery, intense exercise) also promotes ACTH release, regardless of the level of circulating cortisol or the time of day.
Cortisol has multiple effects on intermediate carbohydrate, protein, and fatty acid metabolism, as well as maintenance and regulation of immune and circulatory function. Glucocorticoids enhance gluconeogenesis, elevate blood glucose, and promote hepatic glycogen synthesis. The catabolic effect of glucocorticoids is partially blocked by insulin. The net effect on protein metabolism is enhanced degradation of muscle tissue and negative nitrogen balance. In supraphysiologic amounts, glucocorticoids suppress growth hormone secretion and impair somatic growth. The anti-inflammatory actions of cortisol relate to its effect in stabilizing lysosomes and promoting capillary integrity. Cortisol also antagonizes leukocyte migration inhibition factor, thus reducing white cell adherence to vascular endothelium and diminishing leukocyte response to local inflammation. Phagocytic activity does not decrease, although the killing potential of macrophages and monocytes is diminished. Other diverse actions include the facilitation of free water clearance, maintenance of blood pressure, a weak mineralocorticoid effect, promotion of appetite, stimulation of hematopoiesis, and induction of liver enzymes.
Mineralocorticoid Physiology
Aldosterone is the most potent mineralocorticoid produced by the adrenal gland. This hormone binds to receptors in sweat glands, the alimentary tract, and the distal convoluted tubule of the kidney. Aldosterone is a major regulator of extracellular volume and potassium homeostasis through the resorption of sodium and the secretion of potassium by these tissues. The major regulators of aldosterone release are the renin–angiotensin system and serum potassium (Fig. 46-3). The juxtaglomerular apparatus that surrounds the renal afferent arterioles produces renin in response to decreased perfusion pressures and sympathetic stimulation. Renin splits the hepatic precursor angiotensinogen to form the decapeptide, angiotensin I, which is then altered enzymatically by converting enzyme (primarily in the lung) to form the octapeptide angiotensin II. Angiotensin II is the most potent vasopressor produced in the body. It directly stimulates the adrenal cortex to produce aldosterone. The renin–angiotensin system is the body’s most important protector of volume status. Other stimuli that increase the production of aldosterone include hyperkalemia and, to a limited degree, hyponatremia, prostaglandin E, and ACTH.
Glucocorticoid Excess (Cushing Syndrome)
Cushing syndrome, caused by either overproduction of cortisol by the adrenal cortex or exogenous glucocorticoid therapy, results in a syndrome characterized by truncal obesity, hypertension, hyperglycemia, increased intravascular fluid volume, hypokalemia, fatigability, abdominal striae, osteoporosis, and muscle weakness. Most cases of Cushing syndrome that occur spontaneously are due to bilateral adrenal hyperplasia secondary to ACTH produced by an anterior pituitary microadenoma or nonendocrine tumor (e.g., of the lung, kidney, or pancreas) (see Chapter 36). The primary overproduction of cortisol and other adrenal steroids is caused by an adrenal neoplasm in approximately 20% to 25% of patients with Cushing syndrome. These tumors are usually unilateral, and approximately half are malignant. When Cushing syndrome occurs in patients older than 60 years of age, the most likely cause is an adrenal carcinoma or ectopic ACTH produced from a nonendocrine tumor. Finally, an increasingly common cause of Cushing syndrome is the prolonged administration of exogenous glucocorticoids to treat a variety of illnesses.
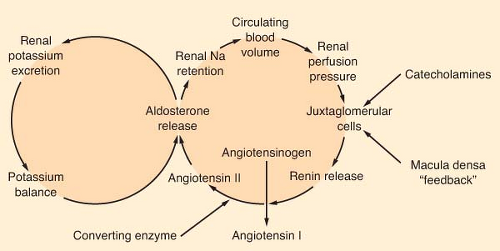 Figure 46-3. Interrelationship of the volume and potassium feedback loops on aldosterone secretion. (Reprinted from Petersdorf RG, ed. Harrison’s principles of internal medicine. 10th ed. New York: McGraw-Hill; 1983, with permission.) |
The signs and symptoms of Cushing syndrome follow from the known actions of glucocorticoids. Truncal obesity and thin extremities reflect increased muscle wasting and a redistribution of fat in facial, cervical, and truncal areas. Impaired calcium absorption and a decrease in bone formation may result in osteopenia. Sixty percent of patients have hyperglycemia, but overt diabetes mellitus (DM) occurs in <20%. Hypertension and fluid retention are seen in most patients. Profound emotional changes, ranging from emotional lability to frank psychosis, may be present. An increased susceptibility to infection reflects the immunosuppressive effects of corticosteroids. Hypokalemic alkalosis without distinctive physical findings is common when adrenal hyperplasia is caused by ectopic ACTH production from a nonendocrine tumor.
The laboratory diagnosis of hyperadrenocorticism is based on a variable elevation in plasma and urinary cortisol levels, urinary 17-hydroxycorticosteroids, and plasma ACTH. Once the diagnosis is established, simultaneous measurement of plasma ACTH and cortisol levels can determine whether the Cushing syndrome is due to primary pituitary or adrenal disease.27
Alternatively, a dexamethasone suppression test can be used. Patients with pituitary adenomas frequently show depression in cortisol and 17-hydroxycorticosteroid levels when a high dose of dexamethasone is administered because the tumor retains some negative-feedback control, while adrenal tumors do not.
Anesthetic Management
General considerations for the preoperative preparation of the patient include treating hypertension and diabetes and normalizing intravascular fluid volume and electrolyte concentrations. Diuresis with the aldosterone antagonist spironolactone helps mobilize fluid and normalize potassium concentration. Careful positioning of the osteopenic patient is important to avoid
fractures. Intraoperative monitoring is planned after evaluation of the patient’s cardiac reserve and consideration of the site and extent of the proposed surgery. When either unilateral or bilateral adrenalectomy is planned, glucocorticoid replacement therapy is initiated at a dose equal to full replacement of adrenal output during periods of extreme stress (see “Steroid Replacement During the Perioperative Period”). The total dosage is reduced by approximately 50% per day until a daily maintenance dose of steroids is achieved (20 to 30 mg/day). Hydrocortisone given in doses of this magnitude exerts significant mineralocorticoid activity, and additional exogenous mineralocorticoid is usually not necessary during the perioperative period. After bilateral adrenalectomy, most patients require 0.05 to 0.1 mg/day of fludrocortisone (9-α-fluorohydrocortisone) starting around day 5 to provide mineralocorticoid activity. Slightly higher doses may be needed if prednisone is used for glucocorticoid maintenance because it has little intrinsic mineralocorticoid activity. The fludrocortisone dose is reduced if congestive heart failure, hypokalemia, or hypertension develops. For the patient with a solitary adrenal adenoma, unilateral adrenalectomy may be followed by normalization of function in the contralateral gland over time. Treatment plans should therefore be individualized, and adjustments in dosage may be necessary. The production of glucocorticoids or ACTH by a neoplasm may not be eliminated if the tumor is unresectable. These patients often need continuous medical therapy with steroid inhibitors such as metyrapone to control their symptoms.
fractures. Intraoperative monitoring is planned after evaluation of the patient’s cardiac reserve and consideration of the site and extent of the proposed surgery. When either unilateral or bilateral adrenalectomy is planned, glucocorticoid replacement therapy is initiated at a dose equal to full replacement of adrenal output during periods of extreme stress (see “Steroid Replacement During the Perioperative Period”). The total dosage is reduced by approximately 50% per day until a daily maintenance dose of steroids is achieved (20 to 30 mg/day). Hydrocortisone given in doses of this magnitude exerts significant mineralocorticoid activity, and additional exogenous mineralocorticoid is usually not necessary during the perioperative period. After bilateral adrenalectomy, most patients require 0.05 to 0.1 mg/day of fludrocortisone (9-α-fluorohydrocortisone) starting around day 5 to provide mineralocorticoid activity. Slightly higher doses may be needed if prednisone is used for glucocorticoid maintenance because it has little intrinsic mineralocorticoid activity. The fludrocortisone dose is reduced if congestive heart failure, hypokalemia, or hypertension develops. For the patient with a solitary adrenal adenoma, unilateral adrenalectomy may be followed by normalization of function in the contralateral gland over time. Treatment plans should therefore be individualized, and adjustments in dosage may be necessary. The production of glucocorticoids or ACTH by a neoplasm may not be eliminated if the tumor is unresectable. These patients often need continuous medical therapy with steroid inhibitors such as metyrapone to control their symptoms.
There are no specific recommendations regarding the use of a particular anesthetic technique or medication in patients with hyperadrenocorticism. When significant skeletal muscle weakness is present, a conservative approach to the use of muscle relaxants is warranted. Etomidate has been used for temporizing medical treatment of severe Cushing syndrome because of its inhibition of steroid synthesis.
Mineralocorticoid Excess
Hypersecretion of the major adrenal mineralocorticoid aldosterone increases the renal tubular exchange of sodium for potassium and hydrogen ions. This leads to hypertension, hypokalemic alkalosis, skeletal muscle weakness, and fatigue. Possibly as many as 1% of unselected hypertensive patients have primary hyperaldosteronism. The increase in renal sodium reabsorption and extracellular volume expansion is partly responsible for the high incidence of diastolic hypertension in these patients. Patients with primary hyperaldosteronism (Conn syndrome) characteristically do not have edema. Secondary aldosteronism results from an elevation in renin production. The diagnosis of primary or secondary hyperaldosteronism should be entertained in the nonedematous hypertensive patient with persistent hypokalemia who is not receiving potassium-wasting diuretics. Hyposecretion of renin that fails to increase appropriately during volume depletion or salt restriction is an important finding in primary aldosteronism. The measurement of plasma renin levels is useful in distinguishing primary from secondary hyperaldosteronism. It is of limited value in differentiating patients with primary aldosteronism from those with other causes of hypertension because renin activity is also suppressed in approximately 25% of patients with essential hypertension.
Anesthetic Considerations
Preoperative preparation for the patient with primary aldosteronism is directed toward restoring the intravascular volume and the electrolyte concentrations to normal. Hypertension and hypokalemia may be controlled by restricting sodium intake and administration of the aldosterone antagonist spironolactone. This diuretic works slowly to produce an increase in potassium levels, with dosages in the range of 25 to 100 mg every 8 hours. Total-body potassium deficits are difficult to estimate and may be in excess of 300 mEq. Whenever possible, potassium should be replaced slowly to allow equilibration between intracellular and extracellular potassium stores. The usual complications of chronic hypertension need to be assessed.
Adrenal Insufficiency (Addison Disease)
The undersecretion of adrenal steroid hormones may develop as the result of a primary inability of the adrenal gland to elaborate sufficient quantities of hormone or as the result of a deficiency in the production of ACTH.
Clinically, primary adrenal insufficiency is usually not apparent until at least 90% of the adrenal cortex has been destroyed. The predominant cause of primary adrenal insufficiency used to be tuberculosis; however, today, the most frequent cause of Addison disease is idiopathic adrenal insufficiency secondary to autoimmune destruction of the gland. Autoimmune destruction of the adrenal cortex causes both a glucocorticoid and a mineralocorticoid deficiency. A variety of other conditions presumed to have an autoimmune pathogenesis may also occur concomitantly with idiopathic Addison disease. Hashimoto thyroiditis in association with autoimmune adrenal insufficiency is termed Schmidt syndrome. Other possible causes of adrenal gland destruction include certain bacterial, fungal, and advanced human immunodeficiency virus infections; metastatic cancer; sepsis; and hemorrhage. Secondary adrenal insufficiency occurs when the anterior pituitary fails to secrete sufficient quantities of ACTH. Pituitary failure may result from tumor, infection, surgical ablation, or radiation therapy. Pituitary surgery may cause transient adrenal insufficiency requiring supplemental glucocorticoids.28
Patients receiving chronic corticosteroid therapy will not generally have frank adrenal insufficiency, but may have hypothalamic-pituitary-adrenal (HPA) suppression and may develop acute adrenal insufficiency during the stress of the perioperative period. Relative adrenal insufficiency is a common finding in critically ill surgical patients with hypotension requiring vasopressors.29
Clinical Presentation
The cardinal symptoms of idiopathic Addison disease include chronic fatigue, muscle weakness, anorexia, weight loss, nausea, vomiting, and diarrhea. Hypotension is almost always encountered in the disease process. Female patients may exhibit decreased axillary and pubic hair growth because of the loss of adrenal androgen secretion. An acute crisis can present as abdominal pain, severe vomiting and diarrhea, hypotension, decreased consciousness, and shock. Diffuse hyperpigmentation occurs in most patients with primary adrenal insufficiency and is secondary to the compensatory increase in ACTH and β-lipotropin. These hormones stimulate an increase in melanocyte production. Mineralocorticoid deficiency is characteristically present in primary adrenal disease; as a result, there is a reduction in urine sodium conservation. Hyperkalemia may be a cause of life-threatening cardiac dysrhythmias. Adrenal insufficiency secondary to pituitary disease is not associated with cutaneous hyperpigmentation or mineralocorticoid deficiency. Salt and water balance is usually maintained unless severe fluid and electrolyte losses overwhelm the subnormal aldosterone secretory capacity. Organic lesions of pituitary origin require a diligent search for coexisting hormone
deficiencies. Acute adrenal insufficiency from inadequate replacement of steroids on chronic steroid therapy is rare and can present as refractory, distributive shock. In critically ill patients, adrenal insufficiency may not present with classic symptoms. The clinical picture may resemble that of sepsis without a source of infection.30 A high degree of suspicion must be maintained if the patient has cardiovascular instability without a defined cause.31,32
deficiencies. Acute adrenal insufficiency from inadequate replacement of steroids on chronic steroid therapy is rare and can present as refractory, distributive shock. In critically ill patients, adrenal insufficiency may not present with classic symptoms. The clinical picture may resemble that of sepsis without a source of infection.30 A high degree of suspicion must be maintained if the patient has cardiovascular instability without a defined cause.31,32
Diagnosis
The patient’s pituitary–adrenal responsiveness should be determined when the diagnosis of primary or secondary adrenal insufficiency is first suspected. Biochemical evidence of impaired adrenal or pituitary secretory reserve unequivocally confirms the diagnosis. Patients who are clinically stable may undergo testing before treatment is initiated. Those believed to have acute adrenal insufficiency should receive immediate therapy.
Plasma cortisol levels are measured before and 30 and 60 minutes after the intravenous administration of 250 μg of synthetic ACTH. There are multiple determinants for adequate adrenal reserve; usually the plasma cortisol rises at least 500 nmol/L 60 minutes after the injection of the synthetic ACTH.33 Patients with adrenal insufficiency usually demonstrate little or no adrenal response.
Treatment and Anesthetic Considerations
Normal adults secrete about 20 mg of cortisol (hydrocortisone) and 0.1 mg of aldosterone per day. Glucocorticoid therapy is usually given twice daily in sufficient dosage to meet physiologic requirements. A typical regimen in the unstressed patient may consist of prednisone, 5 mg in the morning and 2.5 mg in the evening, or hydrocortisone, 20 mg in the morning and 10 mg in the evening. The daily glucocorticoid dosage is typically 50% higher than basal adrenal output to cover the patient for mild stress. Replacement dosages are adjusted in response to the patient’s clinical symptoms or the occurrence of intercurrent illnesses. Mineralocorticoid replacement is also administered on a daily basis; most patients require 0.05 to 0.1 mg/day of fludrocortisone. The mineralocorticoid dose may be reduced if severe hypokalemia, hypertension, or congestive heart failure develops, or it may be increased if postural hypotension is demonstrated.
Secondary adrenal insufficiency often occurs in the presence of multiple hormone deficiencies. A decrease in ACTH production results in the decreased secretion of cortisol and adrenal androgens, but aldosterone control by more dominant mechanisms remains intact. A liberal salt diet is encouraged. Glucocorticoid substitution follows the same guidelines previously outlined for primary adrenal insufficiency.
Immediate therapy of acute adrenal insufficiency is mandatory, regardless of the etiology, and consists of electrolyte resuscitation and steroid replacement (Table 46-6). Initial therapy begins with the rapid intravenous administration of an isotonic crystalloid solution. A dose of 100 mg of hydrocortisone is administered as an intravenous bolus over several minutes. Steroid replacement is continued during the first 24 hours with 100 mg of intravenous hydrocortisone given every 8 hours. If the patient is stable, the steroid dose is reduced starting on the second day. After adequate fluid resuscitation, if the patient continues to be hemodynamically unstable, inotropic support may be necessary. Invasive monitoring is extremely valuable as a guide to both diagnosis and therapy.
Table 46-6. Management of Acute Adrenal Insufficiency | ||
|---|---|---|
|
Steroid Replacement During the Perioperative Period
Perioperatively, patients with adrenal insufficiency and those with HPA suppression from chronic steroid use require additional corticosteroids to mimic the increased output of the normal adrenal gland during stress. The normal adrenal gland can secrete up to 100 mg/m2 of cortisol per day or more during the perioperative period.34 The pituitary–adrenal axis is usually considered to be intact if a plasma cortisol level of >19 μg/dL is measured during acute stress, but there is no precise threshold. The degree of adrenal responsiveness has been correlated with the duration of surgery and the extent of surgical trauma. The mean maximal plasma cortisol level measured during major surgery (colectomy, hip osteotomy) was 47 μg/dL. Minor surgical procedures (herniorrhaphy) resulted in mean maximal plasma cortisol levels of 28 μg/dL. Adrenal activity may also be affected by the anesthetic technique used. Regional anesthesia is effective in postponing the elevation in cortisol levels during surgery of the lower abdomen and extremities.35 Deep general anesthesia may also suppress the elevation of stress hormones such as ACTH and cortisol during the surgical procedure.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


