111 Disorders of Plasma Potassium Concentration
Dyskalemias are common electrolyte disorders in the critical care setting that may predispose a patient to serious cardiac arrhythmias.1 The pathophysiology of these electrolyte disturbances can be more easily understood if examined in the context of the major concept for the transport of potassium ions (K+) across membranes. This process has two components, an open channel for K+ in the cell membrane and a force to cause K+ to move across cell membranes.
 Potassium Channels
Potassium Channels
Clinical Examples
Sulfonylurea drugs stimulate the release of insulin.2 They act by diminishing the open probability of the KATP channels. When fewer K+ ions exit from pancreatic β cells, the ICF voltage becomes less negative. This causes voltage-gated calcium ion (Ca2+) channels to open, and thereby the concentration of Ca2+ in the ICF rises, which provides a signal for the release of insulin from these cells. By virtue of a similar cascade of events, sulfonylurea drugs can cause vasoconstriction by raising the concentration of Ca2+ in the ICF in vascular smooth muscle and hence can be used to improve hemodynamics in patients with septic shock3 (Figure 111-1).
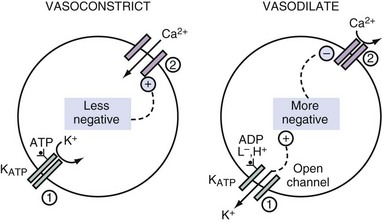
 Driving Forces
Driving Forces
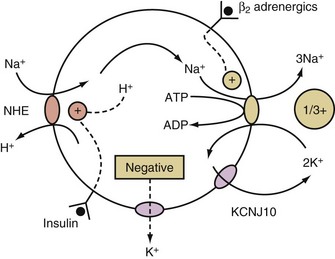
K+ will move into a compartment that has a more negative voltage when K+ channels are open. To create this negative voltage in cells, cations are exported at a faster rate than anions. The cations are usually sodium ions (Na+) because of their abundance and the presence of a means to cause their movement out of cells, the activity of the electrogenic Na+/K+-ATPase. This ion pump is electrogenic because it exports 3 Na+ while importing only 2 K+ (Figure 111-2). Because Na+ movement is not accompanied by movement of ICF anions (because macromolecular phosphates such as RNA, DNA, and phospholipids are impermeable), a negative intracellular voltage is generated. Open KCHJ10 K+ ion channels in the immediate vicinity of the Na+/K+-ATPase in the plasma membrane serve the purpose of providing K+ to the K+ binding site of the Na+/K+-ATPase to permit continuing function of this critical electrogenic system (see Figure 111-2).
 Regulation of Potassium Homeostasis
Regulation of Potassium Homeostasis
Distribution of Potassium Between Extracellular and Intracellular Fluid Compartments
Electroneutral Entry of Sodium into Cells
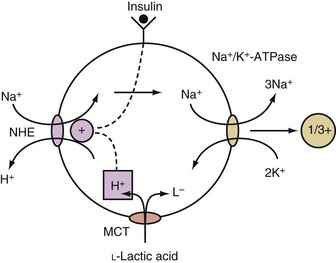
This occurs when Na+ enters cells in exchange for hydrogen ions (H+) via the Na+/H+ exchanger (NHE) (see Figure 111-2).4 The NHE is normally inactive in cell membranes, as can be deduced from the fact that it catalyzes an electroneutral exchange and that the concentrations of its substrates (Na+ in the ECF and H+ in the ICF compartment) are considerably higher than that of its products (Na+ in the ICF and H+ in the ECF compartment) in steady state. The two major activators of NHE are insulin and a higher concentration of H+ in the ICF compartment (Figure 111-2).
Hormones that Affect the Distribution of Potassium
Catecholamines
β2-Adrenergic agonists activate the Na+/K+-ATPase via a cAMP-dependent mechanism that leads to phosphorylation of this ion pump5 and the export of preexisting intracellular Na+. Therefore, hypokalemia may develop in conditions where there is a surge of catecholamines (e.g., patients with a subarachnoid hemorrhage, myocardial ischemia, and/or an extreme degree of anxiety). β2-Agonists may be used to cause a shift of K+ into cells in the emergency treatment of patients with hyperkalemia. On the other hand, non-selective β-blockers have been used in the treatment of patients with thyrotoxic hypokalemic periodic paralysis and are a potential therapy for other conditions of acute hypokalemia due to shift of K+ into cells owing to a surge of catecholamines.
Insulin
The effect of insulin to shift K+ into cells is due primarily to an augmentation of the electroneutral entry of Na+ into cells via NHE.4 This, in conjunction with stimulating the electrogenic Na+/K+-ATPase, causes the voltage in cells to become more negative (see Figure 111-2). This effect of insulin is utilized clinically in the emergency treatment of patients with hyperkalemia.6,7
Acid-Base Influences
Acids That May Cause a Shift of Potassium Into Cells
When an acid is added to the body, most of its H+ are buffered in the ICF compartment.8 Only monocarboxylic acids, however, can enter cells via a specific transporter, and this is an electroneutral process.9 Once a monocarboxylic acid such as L-lactic acid enters cells on this transporter, its H+ are released, and if this occurs in close approximation to NHE in the cell membrane, it becomes activated, and the net result is the electroneutral entry of Na+ into these cells, which causes a rise in their intracellular concentration of Na+. This in turn causes more Na+ and positive voltage to exit from cells. The net result is the generation of a more negative voltage, which causes the retention of K+ in these cells (Figure 111-3).10
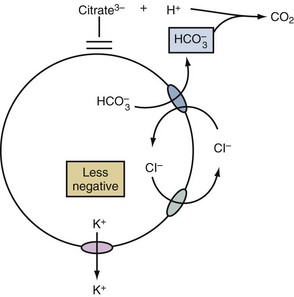
Acids That May Cause a Shift of Potassium Out of Cells
A shift of K+ out of cells may occur in patients with metabolic acidosis due to acids that are not substrates for the monocarboxylic acid transporter (e.g., HCl, citric acid). In this setting, the mechanism begins with the net exit of bicarbonate ions (HCO3−) from cells.11 This exit is an electroneutral process because it occurs on the Cl−/HCO3− anion exchanger (AE) (Figure 111-4). Nevertheless, the process becomes electrogenic because it results in a rise in the concentration of Cl− in the ICF compartment. Since virtually all cells have Cl− channels in their cell membranes,12 the usual negative voltage forces some of these Cl− to exit cells in an electrogenic fashion. As a result of the less negative voltage inside these cells, more K+ will exit.13
Clinical Pearls
Although the addition of inorganic acids (e.g., HCl) causes a shift of K+ out of cells, patients with chronic hyperchloremic metabolic acidosis (e.g., patients with chronic diarrhea or those with renal tubular acidosis [RTA]) usually have a low PK because of excessive loss of K+ in the diarrhea fluid14 or in the urine.15 Although hypokalemia is a common finding in patients with metabolic alkalosis,16 this usually reflects renal K+ wasting for the most part due to the underlying disorder (e.g., vomiting, diuretic use, primary hyperaldosteronism) rather than the small effect of alkalemia to shift K+ into cells. Respiratory acid-base disorders cause only small changes in the PK, because there is little movement of Na+ across cell membranes in these disorders.17
Tissue Anabolism/Catabolism
Hypokalemia may develop in conditions with rapid cell growth if insufficient K+ is given. Examples include the use of total parenteral nutrition (TPN), rapidly growing malignancies, and during treatment of DKA or pernicious anemia. On the other hand, hyperkalemia may be seen in patients with crush injury or tumor lysis syndrome.18 In these patients, factors that compromise the kidney’s ability to excrete K+ are usually present. In patients with DKA, there is total body K+ depletion,19 but hyperkalemia is present because there is a shift of K+ from cells secondary to a lack of insulin. The corollary is that during therapy, complete replacement of the deficit of K+ must await the provision of cellular constituents (phosphate, amino acids, Mg2+, etc.) and the presence of anabolic signals.
 Long-Term Regulation of Potassium Homeostasis
Long-Term Regulation of Potassium Homeostasis
Control of the renal excretion of K+ maintains overall daily K+ balance. Although the usual intake of K+ in adults eating a typical western diet is close to 1 mmol/kg body weight, K+ excretion can decline to a nadir of 10 to 15 mmol/d when there is virtually no K+ intake,20 whereas the rate of excretion of K+ can match an intake of more than 200 mmol/d with only a minor rise in the PK.
Control of K+ excretion occurs primarily in the late distal convoluted tubule up to the end of the cortical collecting duct (the abbreviation CCD will be used in this chapter to indicate all of these nephron segments).19 There are two components that affect the rate of excretion of K+: the flow rate in the CCD and the net secretion of K+ by principal cells in the CCD. It is the latter which adjusts the luminal concentration of K+ ([K+]CCD) and thereby regulates the rate of excretion of K+:
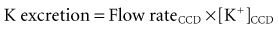
Flow Rate in the LATE CORTICAL DISTAL NEPHRON
When vasopressin acts, the flow rate in the CCD is determined by the rate of delivery of osmoles, because the osmolality of fluid in the terminal CCD is fixed (equal to the plasma osmolality (Posm)21:
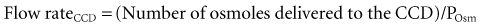
The major osmoles in the lumen of CCD are Na+, Cl−, and urea. Owing to urea recycling within the nephron, almost 75% of osmoles delivered to the CCD are urea (see Reference 22 for more detailed information).
Clinical Example
A patient with HIV and pneumocystis carinii pneumonia is treated with trimethoprim and develops hyperkalemia.23 Because his dietary intake is poor, the rate of delivery of osmoles (mainly urea) to the CCD is low, which means that the flow rate in his CCD is also diminished. This increases the concentration of trimethoprim in the lumen of the CCD (same quantity of trimethoprim is now contained in a smaller volume). Hence the ability of trimethoprim to block epithelial Na+ channels (ENaC) in principal cells in the CCD will be enhanced.24 Furthermore, in the presence of diminished ability to secrete K+ in the CCD owing to a less negative TE luminal voltage, the low flow rate in CCD will further compromise the ability to excrete K+. Increasing the rate of delivery of Na+ and Cl− with a loop diuretic can help augment the rate of excretion of K+ by increasing the flow rate in the CCD. Of greater importance, it will lower the concentration of trimethoprim in the luminal fluid in the CCD, and hence trimethoprim becomes less effective in blocking ENaC.25
Potassium CONCENTRATION in the Lumen of the LATE Cortical DISTAL NEPHRON
The secretory process for K+ in principal cells has two elements. First, a lumen negative voltage must be generated via electrogenic reabsorption of Na+ via ENaC. Actions of aldosterone increase the number of open ENaC. The steps for aldosterone action include its binding to the cytoplasmic aldosterone receptor in principal cells, entry of this hormone-receptor complex into the nucleus, and then the synthesis of new proteins including the serum and glucocorticoid regulated kinase (SGK).26 SGK phosphorylates and inactivates Nedd4-2 (Figure 111-5). As a result, this increases the number of open ENaC units in the luminal membrane of principal cells in the CCD. Second, open K+ channels must be present in the luminal membranes of principal cells in the CCD. K+ channels (ROMK) are abundant and have a high open probability in the absence of hypokalemia, and therefore they do not seem to be rate limiting for net secretion of K+ in most patients.
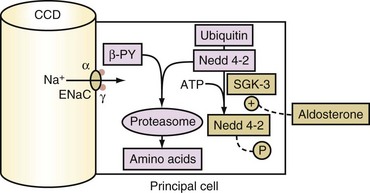
Glucocorticoids do not usually stimulate the secretion of K+ in the CCD because principal cells have a pair of enzymes called 11β-hydroxysteroid dehydrogenase (11β-HSDH). These enzymes convert cortisol to a metabolite (cortisone) that does not bind to the mineralocorticoid receptor (see Figure 111-5). Cortisol, however, can exert a mineralocorticoid effect if the activity of 11β-HSDH is decreased or if it is overwhelmed by an abundance of cortisol.
Under most circumstances, variations in the concentration of Na+ in the luminal fluid in the CCD does not regulate the secretion of K+.27 The reabsorption of Na+ in the CCD can be electroneutral or electrogenic, depending on whether the same quantity of Cl− (electroneutral) or a smaller quantity of Cl− (electrogenic) is reabsorbed as compared to Na+. The pathway(s) for the reabsorption of Cl− in the CCD is (are) not well defined, but it is likely that paracellular pathways play an important role.28,29
Reabsorbing less Cl− than Na+ in the CCD can occur for three reasons, as depicted in Figure 111-6. First, Na+ is delivered to the CCD with little Cl−. A key finding in these patients is a Cl−-poor urine.30 Second, reabsorption of Cl− in the CCD may be inhibited; this mechanism is suspected when the urine is not Cl−-poor. It appears that HCO3− and/or an alkaline luminal pH in the CCD may inhibit Cl− reabsorption31 (see Figure 111-6, middle panel). Third, a greater lumen-negative voltage in the CCD could develop when the delivery of Na+ and Cl− are very high and if the capacity for Cl− reabsorption is less than that for Na+. This requires a stimulated reabsorption of Na+ via ENaC in the CCD (see Figure 111-6, right panel).
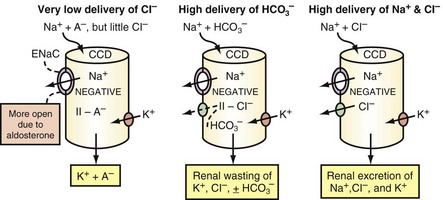
If there are near-equal rates of absorption of Na+ and Cl− in the CCD, an appreciably greater lumen-negative voltage cannot be generated, and hyperkalemia will develop if the intake of K+ remains high.28,32
 Tools to Assess Control of Renal Excretion of Potassium
Tools to Assess Control of Renal Excretion of Potassium
Examine Rate of Excretion Of Potassium
To assess the renal response in a patient with hypokalemia or hyperkalemia, we use the expected rate of K+ excretion when these electrolyte abnormalities are due to nonrenal causes. With a K+ deficit, the expected response is to excrete less than 15 mmol of K+/d.20,45 With a surfeit of K+, the expected response is to excrete greater than 200 mmol/d, values observed in response to a K+ load with a minor increase in PK.33
To assess the rate of excretion of K+, a 24-hour urine collection is not necessary. One can use the UK/UCreatinine ratio in a spot urine sample even though there is a diurnal variation in K+ excretion,21 because creatinine is excreted at a near-constant rate throughout the day.34 Moreover, the UK/UCreatinine in spot urine samples provides more relevant information because it can be evaluated relative to the PK at that time. The expected UK/UCreatinine ratio in a patient with hypokalemia is less than 1 mmol K+/mmol creatinine (less than 10 mmol K+/g creatinine), whereas in a patient with hyperkalemia, the expected UK/UCreatinine ratio is greater than 15 mmol K+/mmol creatinine (greater than 150 mmol K+/g creatinine).
Establish Basis for Abnormal Rate of Excretion of Potassium
In a patient with hypokalemia, a higher than expected rate of excretion of K+ implies that the lumen-negative voltage is abnormally more negative and that open luminal K+ channels (likely ROMK) are present in the luminal membranes of the CCD.39 The greater lumen negative voltage is due to reabsorbing more Na+ than Cl− per unit time in the CCD. The converse is true in a patient with hyperkalemia where there is a lower than expected rate of excretion of K+.
The clinical indices that help in the differential diagnosis of the pathophysiology of the abnormal rate of electrogenic reabsorption of Na+ in CCD are an assessment of the ECF volume and the ability to conserve Na+ and Cl− in response to a contracted effective arterial blood volume. The measurement of the activity of renin (PRenin) and the level of aldosterone in plasma (PAldosterone) are also helpful in this setting (Box 111-1).35
Box 111-1
Plasma Renin and Aldosterone Values to Assess the Basis of Hypokalemia or Hyperkalemia
| Lesions That Cause Hypokalemia | ||
| Renin | Aldosterone | |
| Adrenal Gland | ||
| Primary hyperaldosteronism | Low | High |
| Glucocorticoid remediable hyperaldosteronism | Low | High |
| Kidney | ||
| Renal artery stenosis | High | High |
| Malignant hypertension | High | High |
| Renin-secreting tumor | High | High |
| Liddle’s syndrome | Low | Low |
| Disorders involving 11β-HSDH | Low | Low |
| Lesions That Cause Hyperkalemia | ||
| Adrenal Gland | ||
| Addison’s disease | High | Low |
| Kidney | ||
| Pseudohypoaldosteronism type 1 | High | High |
| Hyporeninemic hypoaldosteronism | Low | Low |
Hyperkalemia
 Therapy of Hyperkalemia
Therapy of Hyperkalemia
Medical Emergencies
The major danger of a severe degree of hyperkalemia is a cardiac arrhythmia. Because mild electrocardiographic (ECG) changes may progress rapidly to a dangerous arrhythmia, any patient with an ECG abnormality related to hyperkalemia should be considered as a medical emergency. We would aggressively treat patients with a PK greater than 7.0 mmol/L, even in the absence of ECG changes—the exceptions include those who develop hyperkalemia after extreme exercise (the super-marathon47).
Induce a Shift of Potassium Into the Intracellular Fluid
Insulin
A number of studies support the use of insulin to treat acute hyperkalemia (reviewed in Reference 6). Large doses of insulin (20 units of regular insulin) are needed to have high enough levels of insulin in plasma for a maximal shift of K+ into cells. Give enough glucose, and monitor PGlucose closely to avoid hypoglycemia.
β2-Adrenergic Agonists
Although a number of studies suggest that β2-agonists (e.g., 20 mg of nebulized albuterol) is effective treatment to lower PK rapidly, we do not use these agents as a primary treatment of emergency hyperkalemia for two reasons. First, in a number of studies it was noted that 20% to 40% of patients with end-stage renal disease are resistant to this therapy, and it is not possible to predict who the non-responders will be. Second, we are concerned about the safety of these drugs in the doses used for the treatment of hyperkalemia; these doses, which are 4 to 8 times those prescribed for the treatment of acute asthma. The combination of nebulized β2-agonists and insulin was reported to produce a greater fall in PK (1.2 mmol/L) compared with either drug alone (~0.65 mmol/L).48 One should note, however, that only 10 units of regular insulin were given in this study, and the magnitude of the fall in PK was lower than that observed in other studies using higher doses of insulin.49 Thus, it remains uncertain whether β2-agonists have a PK-lowering effect that is additive to that of higher doses of insulin.
Sodium Bicarbonate
A number of studies have found NaHCO3 therapy to be ineffective, as the sole treatment of hyperkalemia.49–51 Notwithstanding, these studies were performed in stable hemodialysis patients who did not have significant acidemia. Studies that examined the combined use of NaHCO3 with insulin also have yielded conflicting results.52,53 Thus the question remains, Would NaHCO3 be effective in patients with a more significant degree of acidemia? There are no data in the literature to answer this question definitively (for review, see Reference 6). Given this uncertainty, we only use NaHCO3 in addition to other therapies to treat emergency hyperkalemia in patients with a significant degree of acidemia. Caution is warranted because an excessive administration of NaHCO3 has the risk of inducing hypernatremia, ECF volume expansion, carbon dioxide retention, and acute hypocalcemia.
 Clinical Approach
Clinical Approach
It is imperative to recognize when hyperkalemia represents a medical emergency because therapy must take precedence over diagnosis (Figure 111-7). A step-by-step approach to diagnosis of hyperkalemia is illustrated in Figures 111-8 and 111-9.
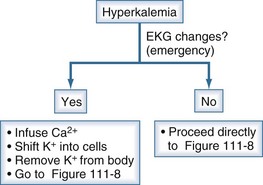
Figure 111-7 Initial clinical approach for the patient with hyperkalemia.
(From Halperin ML. The ACID truth and BASIC facts—with a sweet touch, an enLYTEnment. 5th ed. Toronto: RossMark Medical Publishers; 2003. Reproduced with permission ref 74.)
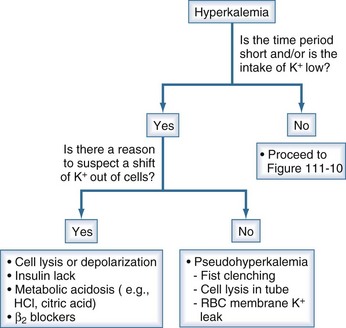
Figure 111-8 Initial steps in the clinical diagnosis of the cause of hyperkalemia.
The most important issue is to determine if a shift of K+ out of cells is likely; this is done by assessing the time course for the rise in the PK and whether there was little intake of K+. If that was the case and there is no reason to suspect a shift of K+ out of cells, pseudohyperkalemia should be ruled out. In this latter setting, there should not be ECG changes related to hyperkalemia. If this is ruled out, proceed to Figure 111-9, and examine the rate of excretion of K+.
(From Halperin ML. The ACID truth and BASIC facts—with a sweet touch, an enLYTEnment. 5th ed. Toronto: RossMark Medical Publishers; 2003. Reproduced with permission ref 74.)
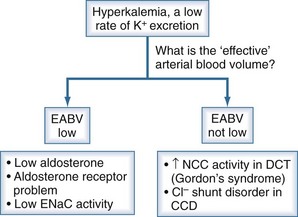
Figure 111-9 Basis for the low rate of excretion of K+.
(From Halperin ML. The ACID truth and BASIC facts—with a sweet touch, an enLYTEnment. 5th ed. Toronto: RossMark Medical Publishers; 2003. Reproduced with permission, ref 74.)
In Box 111-2, we provide a list of causes of hyperkalemia and of hypokalemia based on the presence or absence of hypertension.
Box 111-2
Dyskalemias and Blood Pressure*
Hyperkalemia
Hypokalemia


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


