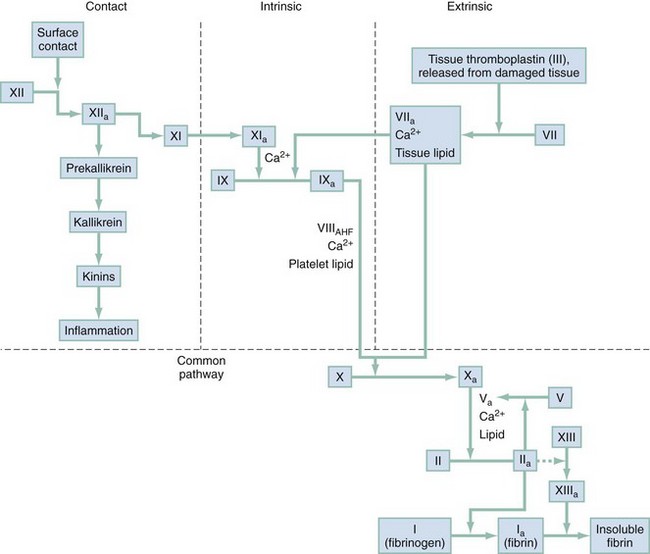
Chapter 122 Vascular integrity is maintained by a lining of nonreactive overlapping endothelial cells supported by a basement membrane, connective tissue, and smooth muscle. These cells are important in maintaining a barrier to macromolecules and, when injured, in contributing to the metabolic response and local vasoconstriction. The vascular endothelium is an important contributor to hemostasis.1 Platelets have multiple and ever-expanding roles in our understanding of hemostasis. They are complex cytoplasmic fragments released from bone marrow megakaryocytes under the control of thrombopoietin. Platelets contain lysosomes, granules, a trilaminar plasma membrane, microtubules, and a canalicular system. Granules are an important component of hemostasis and contain platelet factor 4, adhesive and aggregation glycoproteins, coagulation factors, and fibrinolytic inhibitors. Each participates in the process of coagulation. The platelet’s role is termed primary hemostasis, and it serves as the initial defense against blood loss.1,2 A fibrin clot that incorporates coagulation factors usually reinforces a platelet clot. Platelet activity is summarized in Box 122-1. Any of the steps listed may be absent, altered, or inhibited by inherited or acquired disorders. The coagulation pathway is a complex system of checks and balances that results in controlled formation of a fibrin clot. Coagulation factors have been given standard Roman numerals matching their order of discovery (Box 122-2). A simplified version of the coagulation pathway is presented in Figure 122-1. The clotting cascade is traditionally depicted as consisting of intrinsic and extrinsic pathways. The intrinsic pathway is initiated by exposure of blood to a negatively charged surface, such as a glass surface in the activated partial thromboplastin clotting time. The extrinsic pathway is activated by tissue factor exposed at the site of vessel injury or thromboplastin. Both pathways converge to activate factor X, which then activates prothrombin to thrombin. The primary physiologic event that initiates clotting is exposure of tissue factor at the injured vessel site. Tissue factor is a critical cofactor that is required for activation of factor VII. Activated factor VII activates factor X directly as well as indirectly by activating factor IX. Because of limited amounts of tissue factor and rapid inactivation by tissue factor pathway inhibitor, the extrinsic pathway initiates the clot process. Sustained generation of thrombin and clot formation depend on the intrinsic pathway through activation of factor IX by activated factor VII, which helps explain the bleeding problems associated with hemophilia.3 Intrinsic, extrinsic, and common pathways function normally for hemostasis to occur, and each may be evaluated with laboratory tests. The clinically important groups of coagulation factors are as follows: Thrombin-sensitive factors contributing to the metabolic response and local vasoconstriction: I, V, VIII, XIII Vitamin K-sensitive factors: II, VII, IX, X Sites of heparin activity: IIa, IXa, Xa (major site), XIa, platelet factor 3 Removal and dilution of activated clotting factors through blood flow, which also mechanically opposes growth of the hemostatic plug Modulation of platelet activity by endothelium-generated nitric oxide and prostacyclin Removal of activated coagulation components by the reticuloendothelial system Regulation of the clotting cascade by antithrombin III, protein C, protein S, and tissue factor pathway inhibitor An outline of the history and physical examination is presented in Box 122-3. The history alone may be useful in differentiating between platelet and coagulation factor abnormalities. Platelet disorders are usually manifested as acquired petechiae, purpura, or mucosal bleeding and are more common in women. Coagulation problems are commonly congenital, are characterized by delayed deep muscle or joint bleeding, and are seen more often in men. A definitive diagnosis depends on laboratory evaluation. Tests pertinent to the ED are discussed in the following sections and listed in Box 122-4. The complete blood count assesses the degree of anemia associated with the bleeding episode. Reductions in hemoglobin and hematocrit often lag behind the actual loss of red blood cells (RBCs) in acute hemorrhage because of a slow equilibration time. The peripheral blood smear may demonstrate schistocytes or fragmented RBCs in disseminated intravascular coagulation (DIC). Teardrop-shaped or nucleated RBCs may reflect myelophthisic disease. A characteristic white blood cell morphologic condition is seen with thrombocytopenia associated with infectious mononucleosis, folate or vitamin B12 deficiency, or leukemia.4 Vascular disorders have signs and symptoms similar to those of thrombocytopenic states. The inherited forms are rare. Acquired forms are usually associated with connective tissue changes or endothelial damage. The differential diagnosis of vascular disorders is listed in Box 122-5. Platelet abnormalities can be caused by congenital disorders, but most are from acquired conditions.5 The bleeding source is usually capillary, with resultant cutaneous and mucosal petechiae or ecchymosis. Epistaxis, menorrhagia, and gastrointestinal bleeding are common initial symptoms. The bleeding is generally mild and occurs immediately after surgery or dental extractions. Petechiae and purpura may be noted on physical examination, and superficial ecchymoses may be found around a venipuncture site. The purpura associated with platelet disorders is typically asymptomatic and not palpable. This is in contrast to purpura associated with vasculitis, which can burn or itch and is palpable.6 Deep muscle hematomas and hemarthroses are not aspects of the clinical picture. The bleeding time is prolonged, and the platelet count may be low, normal, or high. The differential diagnosis of platelet disorders is listed in Box 122-6. Decreased Production.: Thrombocytopenia from decreased bone marrow production is usually caused by the effects of chemotherapeutic drugs, myelophthisic disease, or direct bone marrow effects of agents such as alcohol or thiazides. Splenic Sequestration.: Splenic sequestration is rare. It is seen primarily with hypersplenism resulting from hematologic malignant disease, portal hypertension, or disorders involving increased splenic RBC destruction, such as hereditary spherocytosis or autoimmune hemolytic anemia. Immune Thrombocytopenia.: Thrombocytopenia associated with increased peripheral destruction of platelets and shortened platelet survival caused by an antiplatelet antibody is seen in a number of diseases. In most cases a cause is identifiable. Low-molecular-weight heparin may be associated with less thrombocytopenia than standard, unfractionated heparin is; however, both forms of heparin demonstrate cross-reactivity.7 Heparin-induced thrombocytopenia (HIT) is a serious immune-mediated side effect associated with heparin.8,9 A meta-analysis including 7287 patients determined the incidence of HIT to be 2.6% for unfractionated heparin and 0.2% for low-molecular-weight heaprin.10 It usually occurs 5 to 7 days after the initiation of heparin treatment. Thrombus develops in approximately half the patients with HIT. The thrombotic complications can lead to loss of a limb in up to 20% and death in as many as 30%. The diagnosis is suggested in the presence of absolute thrombocytopenia or a greater than 50% reduction in platelets after the initiation of heparin. The most specific diagnostic tests for HIT are serotonin release assays, heparin-induced platelet aggregation assays, and solid-phase immunoassays. Platelet-associated IgG levels are commonly elevated, but this finding is less specific or sensitive than the other diagnostic tests. More concerning to the emergency physician is delayed-onset HIT. This form of HIT occurs a median of 14 days after the initiation of heparin, but it has been reported to occur up to 40 days after heparin is started.10 Arterial or venous thrombosis typically develops in patients with HIT after they receive heparin. The administration of heparin can result in the development of antibodies to the heparin and platelet factor 4 complex. The heparin–platelet factor 4–antibody complex is removed from the circulation, resulting in thrombocytopenia; however, this complex also results in the generation of microparticles that have procoagulant properties, which can lead to thrombus formation. Treatment of thrombotic complications in these patients involves the use of direct thrombin inhibitors (lepirudin, argatroban), factor Xa inhibitors (fondaparinux), or heparinoids (danaparoid).8 Digitoxin, sulfonamides, phenytoin, and aspirin are other drugs that may be associated with a thrombocytopenia. The patient has usually ingested the medication within 24 hours. An idiopathic thrombocytopenic purpura type of syndrome has been reported in intravenous cocaine users.11 Clinical trials with platelet glycoprotein IIb/IIIa antagonists suggest that intravenous glycoprotein IIb/IIIa inhibitors may confer an increased risk for associated thrombocytopenia, independent of heparin therapy.12 The platelet count may fall below 10,000/mm3 and be complicated by serious bleeding. Laboratory testing may confirm the presence of antibody, especially with the use of quinine and quinidine. After administration of the drug is stopped, the platelet count improves slowly during a period of 3 to 7 days. A short course of corticosteroid therapy, such as prednisone in a dose of 1 mg/kg with rapid tapering, may facilitate recovery.13,14 Postinfectious immune thrombocytopenia is usually associated with viral diseases such as rubella, rubeola, and varicella. Although many cases associated with sepsis have a mechanical origin, some immune mechanisms have been demonstrated.13 Post-transfusion thrombocytopenia is a rare disorder that causes a precipitous fall in platelets approximately 1 week after the transfusion. In 90% of cases, its origin is linked to the 98% of the population carrying a PLA1 antigen on platelets. Despite the fact that 2% of blood recipients are mismatched with respect to this antigen, it is fortunately a rare occurrence. On transfusion into a PLA1 antigen–negative patient, the platelets with attached PLA1 antibodies provoke an anamnestic response, but the actual mechanism of platelet destruction remains unknown. The platelet count often falls precipitously below 10,000/mm3, with a significant risk for major bleeding. Intracranial hemorrhage occurs in approximately 10% of such cases. Patients are usually middle-aged women with a history of pregnancy who may have been previously sensitized to the PLA1 antigen during pregnancy. Plasma exchange therapy is an effective intervention.13,15
Disorders of Hemostasis
Pathophysiology
Vasculature
Platelets
Coagulation Pathway
Coagulation Control
Clinical Features
History and Physical Examination
Ancillary Evaluation
Complete Blood Count and Blood Smear
Differential Diagnosis and Management
Vascular Disorders
Platelet Disorders
Thrombocytopenia
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Disorders of Hemostasis
Only gold members can continue reading. Log In or Register to continue




