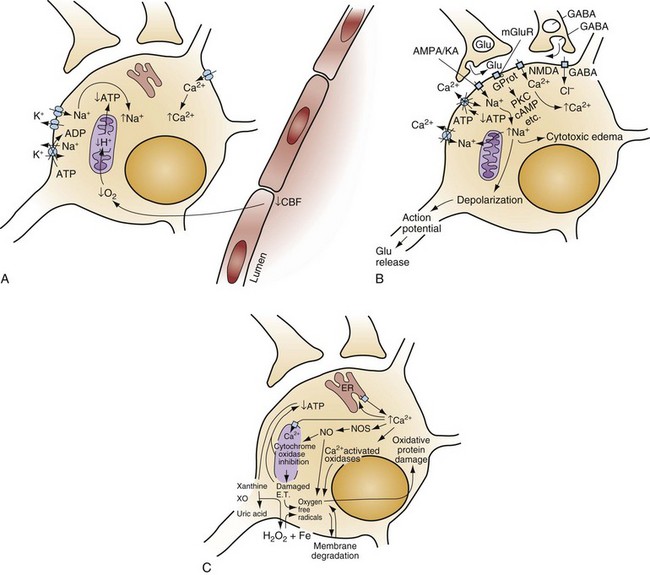
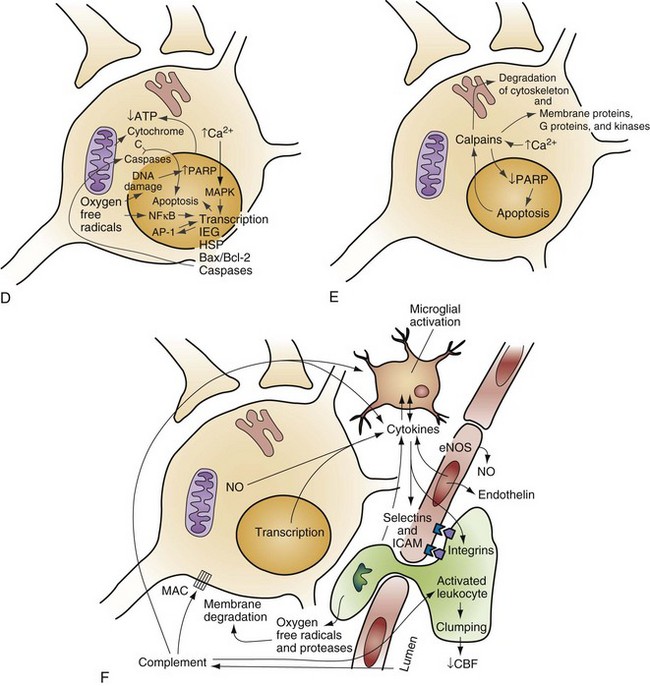
Chapter 8 Recognition of the dominant role of the brain in determining the quality of human life dates back to the dawn of recorded medical history. Until recently, however, medical efforts after cardiac arrest have focused exclusively on cardiac resuscitation. Recent advances in the understanding of the pathophysiologic mechanisms of brain ischemia have encouraged attention to cerebral resuscitation as a critical component of what is now recognized as a complex multisystem pathophysiologic state called post–cardiac arrest syndrome.1 This chapter reviews the pathophysiology of postischemic encephalopathy and discusses therapies for improving neurologic recovery after cardiac arrest. When the brain is deprived of adequate blood flow, the resulting ischemia is characterized by a bewildering array of inter-related physiologic and cellular responses that ultimately result in neuronal cell death (Fig. 8-1).2,3 Although this complex cascade of events can be triggered by periods of ischemia lasting only a few minutes, the resulting neuronal death is usually delayed by hours or days. Furthermore, the biology of cerebral cell death after global cerebral ischemia follows (with slight variations) the pattern of delayed cerebral cell death that follows stroke, traumatic brain injury, and other forms of hypoxic or toxic brain injury. Increased understanding of the brain’s response to injury during the period between insult and neuronal cell death will eventually allow more specific brain resuscitation therapies. Standard management of cardiac arrest and subsequent ischemic brain damage involves restoring cerebral blood flow (CBF) and preventing secondary insult. These treatments have generally not been studied in prospective, randomized controlled trials, but they are supported by clinical experience and limited experimental data. Although proposed and experimental therapies are generally aimed at specific molecular interventions in the pathophysiology of ischemic brain injuries, none of these as yet have proven effective in clinical trials. The most comprehensive review and consensus guideline statement on care of patients with post–cardiac arrest syndrome was produced by the International Liaison Committee on Resuscitation and its constituent bodies, with the endorsement of the American College of Emergency Physicians, Society for Academic Emergency Medicine, Society of Critical Care Medicine, and Neurocritical Care Society.1 Improvements in post–cardiac arrest care, through an inclusive multisystem approach, can increase the likelihood of meaningful recovery in these patients. Implementation of standardized protocols for postresuscitation care that include many or all of the following components have demonstrated increases in survival with a favorable neurologic outcome of up to 27 to 30% in repeated (although poorly controlled) before-and-after studies.4,5 Return of Spontaneous Circulation The efficacy of closed-chest CPR in generating adequate cerebral perfusion is somewhat controversial. Cardiac output during optimal standard closed-chest CPR has previously been estimated to be only 20 to 30% of normal, but more recent data suggest that higher cardiac outputs are possible in clinical practice, and, unquestionably, effective CPR is essential to neurologic recovery after cardiac arrest. Considerable effort has been directed toward the investigation of improved CPR techniques that will prove to be even more effective and for longer periods (see Chapter 9). Hypotension in the postarrest period can dangerously lower cerebral perfusion pressure. Although CBF is normally independent of perfusion pressure over a wide range of arterial blood pressure, such autoregulation is often lost in the injured brain. As a result, perfusion of ischemic tissue becomes passively dependent on arterial pressure, and hypotension can compromise CBF and result in significant additional brain damage.6 Therefore, after return of spontaneous circulation (ROSC), low arterial pressures should be rapidly normalized, with intravascular volume administration and vasopressors used as needed. Because elevated arterial pressures may be needed to provide sufficient CBF, hypertension usually should not be treated in the postresuscitation period. Very high blood pressures may require treatment, but specific cutoffs are controversial. In general, diastolic pressures may be allowed to run as high as 120 mm Hg without requiring treatment. In fact, hypertension is sometimes induced clinically or experimentally with vasopressors in an attempt to raise cerebral perfusion pressure and improve neurologic recovery.6 Because it is unproved, and because risks of this therapy include blood-brain barrier disruption and worsening of vasogenic edema, induced hypertension is not currently a standard therapy. CVR after resuscitation from cardiac arrest is another determinant of CBF and may be affected by hyperventilation and microvascular patency. Although the cerebral circulation may lose its ability to adjust to blood pressure changes after ischemia, attenuated responsiveness to carbon dioxide and oxygen levels in arterial blood can still be present.7 Carbon dioxide is a potent vasoactive agent, and lowering of the arterial carbon dioxide partial pressure (PaCO2) by hyperventilation results in rapid reduction of CBF. Because reductions in CBF reduce total cerebral blood volume, hyperventilation may transiently abort brainstem herniation in the presence of critically elevated intracranial pressure (ICP) until osmotherapy or ventriculostomy can be initiated. When ICP is not elevated, however, the vasoconstriction and increased CVR caused by hyperventilation can cause potentially dangerous reductions in CBF.7 Although controversy surrounds whether increases in ICP are clinically significant after global ischemia, the measurement of ICP is generally not recommended in the management of adult cardiac arrest survivors.8 In general, ventilation to maintain a PaCO2 of 35 to 40 mm Hg is safe and appropriate, and inadvertent hyperventilation should be avoided. CVR may also be elevated after cardiac arrest by endothelin-induced vasospasm or microvascular occlusion by leukocyte clumping or coagulation. Hemodilution, anticoagulation, and antiplatelet agents have been studied in animals for effectiveness in mitigating microvascular occlusion with mostly negative results.9 Acute use of these agents to improve microcirculatory flow has not been studied in human cerebral ischemia. Normal arterial oxygen saturation should be maintained after resuscitation from cardiac arrest. Because the injured brain may not be able to compensate for hypoxia by augmenting CBF, cerebral oxygen delivery may diminish rapidly as the oxygen content of blood decreases. Hyperoxia secondary to the use of 100% oxygen in the immediate postarrest period, however, has also been shown to increase oxidative brain injury in animal models of cardiac arrest and resuscitation.10 Normoxia or mild hyperoxia (arterial partial pressure of oxygen [PaO2] of 80-120 mm Hg with oxyhemoglobin saturation percentage maintained in the high nineties) should be maintained through use of the lowest fraction of inspired oxygen (FIO2) possible. The use of 100% oxygen is appropriate during cardiac arrest, but FIO2 should be titrated downward shortly after the ROSC. A large multicenter cohort study found hyperoxia (defined as PaO2 300 mm Hg or above on first intensive care unit [ICU]–obtained arterial blood gas [ABG] measurement) to be associated with an 18% (95% confidence interval [CI] 14-22%) higher absolute mortality.11 An important clinical trial to compare earlier use of normoxia after ROSC with the more typical prolonged period of postresuscitative hyperoxia has been proposed. Because hypoxia, hypocapnia, and hypercapnia must be avoided, controlled ventilation is appropriate in the period after resuscitation, with muscle relaxation and sedation if needed. Hyperthermia (or fever) exacerbates brain injury and worsens neurologic outcome.12,13 Elevated body temperature increases cerebral metabolic demand by 8 to 13% per degree Celsius, escalates glutamate release, increases oxygen free radical production, and increases cytoskeletal and blood-brain barrier breakdown with increased vasogenic edema.14 Core body temperature (usually rectal, bladder, or esophageal) should be accurately measured in patients resuscitated from cerebral ischemia.15 Hyperthermia may be treated with antipyretics, circulating air or water cooling systems, or evaporative cooling via water mist and fans.15 Aggressive treatment should, at a minimum, be used to prevent temperature increases in the postischemic period in all patients, and the practice of inducing therapeutic hypothermia has emerged as a therapy for comatose survivors of cardiac arrest.
Brain Resuscitation
Perspective
Pathophysiology
Management
Standard Strategies
Treatment of Hypotension, Hypoperfusion, and Hypoxia
Maintenance of Body Temperature
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Brain Resuscitation
Only gold members can continue reading. Log In or Register to continue




