Chapter 48 Diseases of Pulmonary Circulation
Early referral to expert centers is crucial to patient survival.
Developmental Pulmonary Vascular Anatomy
Embryology
Distinctions can be recognized between the proximal and distal pulmonary vasculature both in terms of their embryonic origins and the morphogenetic processes by which they develop. The sixth branchial arch is the embryonic origin of the proximal pulmonary vasculature, and it develops by the process of vasculogenesis. Vasculogenesis is the differentiation and segregation of angioblasts within the mesenchyme, which forms early vascular channels (arteries, veins, and lymphatics), depending on local influences from the epithelium and the mesenchyme. The lung mesenchyme is the embryonic origin of the distal vasculature, which develops by angiogenesis. Angiogenesis is the formation of new vessels from preexisting vascular channels by proliferation and migration of endothelial cells at the tips, which form multiple capillary sprouts. Abnormal maturation or maturational arrest in pulmonary arterial development is reflected in functional derangement that can appear in the newborn period. Persistent pulmonary hypertension of the newborn (PPHN) has been reported in association with pulmonary arterial maturational arrest at week 5 of gestation.1 Normal growth and development of the pulmonary circulation in utero is critical for achieving successful transition to postnatal life.
Multiple factors influence development and growth of pulmonary vasculature in utero, including growth factors. Growth factors such as vascular endothelial growth factor2 and fibroblast growth factor3 appear to be responsible for orderly growth and branching morphogenesis of blood vessels. Quantity, timing, and location are critical determinants of the net effects of these agents. The mechanical stress that endothelial cells must withstand may be the predominant force responsible for development of large vessels after onset of circulation.4
Vascular Smooth Muscle
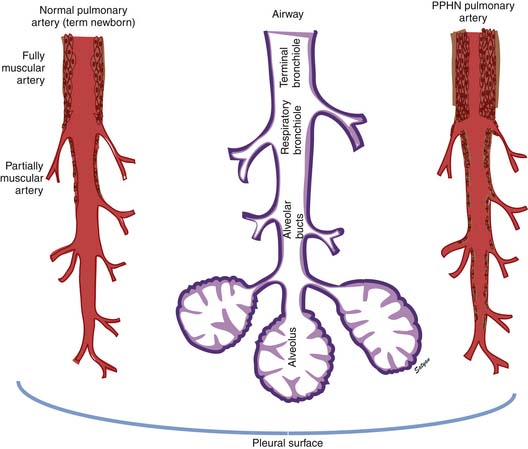
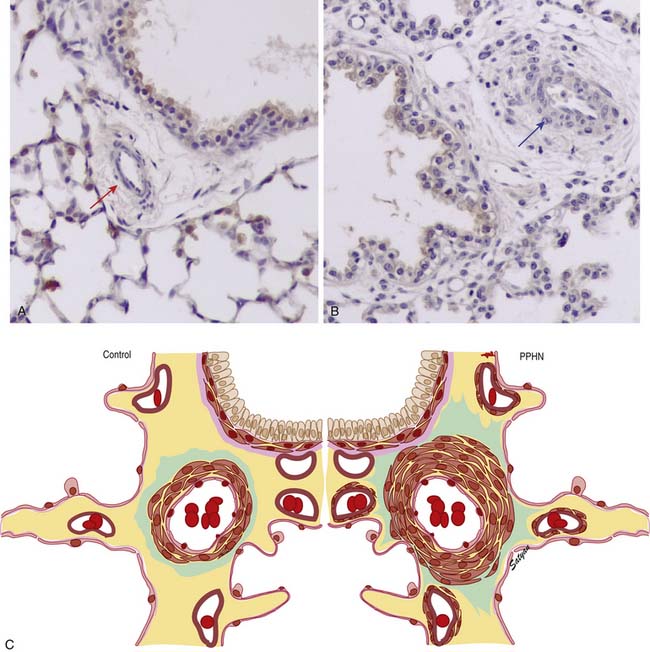
In the normal fetal and term lung, fully muscularized thick-walled preacinar arteries extend to the level of terminal bronchioles, whereas the intra-acinar arteries (i.e., those accompanying respiratory bronchioles) are partially muscular (surrounded by a spiral of muscle) or nonmuscular. Arteries at alveolar ducts and alveolar walls are nonmuscular. Preacinar arteries in the fetus late in gestation and in the newborn have thicker coats of smooth muscle relative to the arterial diameter than do similar arteries in adults, although the fetus actually has less distal extension of smooth muscle in smaller arteries than do older children or adults.5 Figure 48-1 shows the diagrammatic representation of the extension of arterial smooth muscle within the acinus in a normal term newborn and in infants with severe PPHN.6 An increase in intrauterine vascular smooth muscle, resulting from peripheral extension of smooth muscle into vessels that do not normally contain muscle layers, may contribute to the pathophysiology of PPHN7 (see Figure 48-1). Increased muscularization of the pulmonary arteries has been described in infants with severe meconium aspiration syndrome (MAS) with PPHN.8 Neonates who die of PPHN may have a striking distal extension of smooth muscle in the intra-acinar region, thickening of the media and adventitia, and excessive accumulation of the matrix protein in the pulmonary vessels.9 The increase in vascular smooth muscle and its peripheral extension can occur either prenatally or postnatally. In utero ductal ligation 1 to 2 weeks prior to delivery results in severe PPHN at birth associated with distal extension of vascular smooth muscle in newborn lambs10 (Figure 48-2) similar to the changes seen in human infants dying with severe PPHN.
Developmental Pulmonary Vascular Physiology
Hemodynamic Features of Fetal Circulation
Oxygenated blood (PaO2 approximately 30 to 40 mm Hg) returning from the placenta in the umbilical vein11 splits in the liver with slightly more than half passing through the ductus venosus to the inferior vena cava (IVC). The oxygenated blood streams along the medial aspect of the IVC as it enters the right atrium (Figure 48-3). Approximately two thirds of the IVC flow is directed toward the foramen ovale by the eustachian valve and the septum primum and enters the left atrium. The remaining third of the IVC flow mixes with the blood from the superior vena cava and enters the right ventricle. The majority of the right ventricular (RV) output enters the ductus arteriosus and the descending aorta. A small portion enters the lungs via the pulmonary arteries.12 The ratio of blood flow to the pulmonary arteries to the flow that traverses the ductus arteriosus is determined by the fetal pulmonary vascular resistance (PVR).
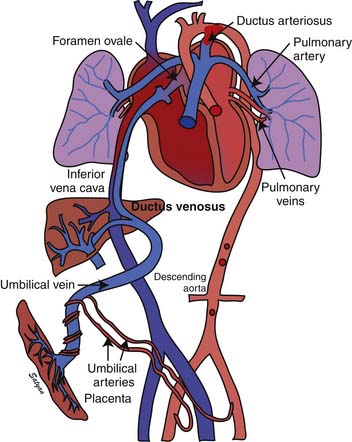
Figure 48–3 Fetal circulation showing the shunting of blood at the ductus venosus, foramen ovale, and ductus arteriosus.
(Copyright Satyan Lakshminrusimha.)
The fetal pulmonary circulation fulfills a unique function. Close to term, fetal lungs in the lamb receive about 5% to 10% of combined ventricular output to meet the metabolic demands of an actively growing organ. More recent data in human fetuses using Doppler echocardiography suggest that approximately 25% of combined ventricular output may enter the lungs near term.13 The presence of large fetal shunts, the foramen ovale and the ductus arteriosus (see Figure 48-3), allows the fetal lung to regulate the amount of blood flow it receives by active vasoconstriction. The distribution of combined ventricular output, measured in fetal lambs ranging from 60 to 150 days gestational age, indicates that the lungs receive only about 3.5% of total output at 0.4 term, but the fraction increases to almost 8% at term.14 This remarkable increase in fetal pulmonary perfusion presumably is related to growth of the fetal lung. The lung increases in weight by sixfold from midgestation to term, and the number of pulmonary vessels increases more than tenfold.15,16 Thus the tremendous increase in cross-sectional area of pulmonary vasculature permits pulmonary blood flow (Qp) to increase throughout gestation. However, Qp remains relatively constant when corrected for wet lung weight with advancing gestation.17 A gradual increase in pulmonary arterial pressure (PAP), accompanied by a relatively constant flow per unit lung tissue, results in increasing fetal PVR with advancing gestation.17 A major part of this increase comes from elevated pulmonary vascular tone associated with low PO2 in the fetal lung.18,19 Maintaining high PVR in utero is important because the function of gas exchange is performed not by the lungs but by the placenta.
Regulation of Pulmonary Vascular Tone in Utero
Low oxygen tension and various mediators play a crucial role in maintaining elevated fetal PVR.20 Vasoconstriction in response to low oxygen tension contributes to high PVR in the fetal lamb as it approaches term.18,19 Decreasing oxygen tension in fetuses at 103 to 104 days of gestation does not increase PVR, but it doubles resistance in fetuses at 132 to 138 days. Conversely, increasing oxygen tension does not change PVR before 100 days of gestation, but it decreases resistance markedly and increases blood flow to normal newborn levels at 135 days of gestation.17
Arachidonic acid metabolites are some of the most powerful vasoconstrictors known. Prostaglandin (PG) F2α and thromboxane A2 (TXA2) are synthesized by the fetal lung via the cyclooxygenase pathway21 and are pulmonary vasoconstrictors in the fetal and newborn lungs. However, TXA2 does not appear to be responsible for maintenance of high PVR in the fetus.22 Blocking PG or thromboxane synthesis does not decrease fetal PVR but prevents the decrease in PVR in response to rhythmic distension of the lung in fetal lambs.23 The cytochrome P450 metabolism of arachidonic acid results in the formation of epoxyeicosatrienoic acids, dihydroxyeicosatetraenoic acids, and 20-hydroxyeicosatetraenoic (HETE) acids. Of these compounds, HETE acids have been shown to constrict pulmonary circulation in newborn piglets.24 However, inhibition of 20-HETE did not reduce basal PVR in fetal lambs.25 Thus it does not appear that cyclooxygenase and CYP450 metabolites of arachidonic acid cause the high vascular tone of the fetal lung.
Leukotrienes are formed from arachidonic acid through the 5-lipooxygenase pathways. Lipoxygenase activity has been demonstrated in human fetal lung as early as 12 to 18 weeks of gestation.26 Leukotriene inhibition has been shown to decrease PVR in fetal lambs by 45%.27 It also reverses hypoxic pulmonary vasoconstriction in newborn lambs but not in newborn piglets.28,29 The specific role of leukotrienes in maintaining high PVR in immature fetuses is unclear. It is possible that they play a role in pathologic states such as hypoxia or inflammation.
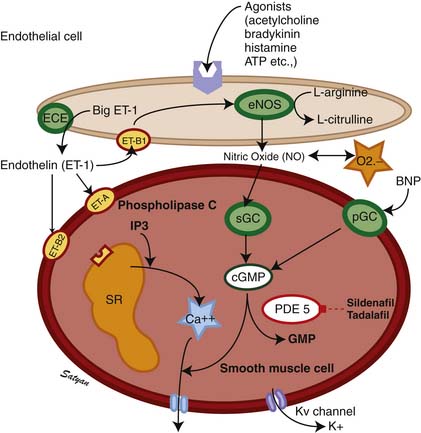
Endothelins (ETs) are 21-residue peptides30,31 whose role in regulating vascular tone and vasomotor responses has been studied intensively in the past decade. Three distinct ET isoforms have been described: endothelin-1 (ET-1), endothelin-2 (ET-2), and endothelin-3 (ET-3), cleaved from ET precursors big ET-1, big ET-2, and big ET-3, respectively, by an ET-converting enzyme. ET-1 synthesized by vascular endothelial cells is a potent vasoconstrictor,32 and its effects in both animal and human studies vary with the tone of the pulmonary vessels, dose of ET-1, and the maturation of vessels.33,34 Of the ETs, ET-1 is the best characterized, and its actions of fetal pulmonary circulation are best studied. Both ET-1 and ET-2 have been shown to dilate the fetal (normally high tone) pulmonary vasculature and constrict the bed when the tone is reduced by ventilation.35 Thus it appears that the response of the pulmonary vasculature to ETs is tone-dependent. In contrast, infusion of big ET-1, the precursor of ET-1, into fetal lambs causes sustained pulmonary vasoconstriction,36 suggesting that this might be the predominant effect of endogenous ET-1.37 Currently at least two receptor subtypes, ETA and ETB, are thought to mediate responses to ETs (Figure 48-4). The ETB receptor plays a role in vasodilation and the ETA receptor plays a role in vasoconstriction. Selective blockade of the ETA receptor causes fetal pulmonary vasodilation.38–40 Some investigators suggest a significant role for an endothelial ETB receptor in vasodilation and a smooth muscle ETB receptor and ETA receptor in vasoconstriction.41,42 Vasoconstriction induced by ET-1 is mediated by calcium,43 whereas the vasodilator properties are mediated by endothelium-derived nitric oxide (NO).42,44 ET-1 may play a role in the change that occurs in the pulmonary vasculature at birth. It is possible that prenatally, endogenous ET-1 primarily stimulates ETA receptors to cause vasoconstriction and that ETB receptors are less active in fetal life.38 However, ETB receptors mediate the vasodilator responses to ET-1 in the fetus, and there is a suggestion that an abundance of ETB receptors may be of physiologic importance in decreasing PVR at birth.40
Transitional Circulation
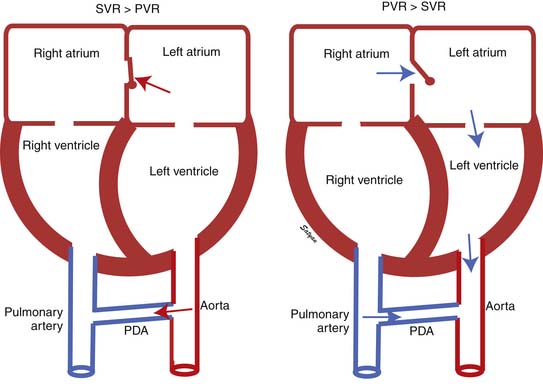
The first stage of transitional circulation is essentially a fetal pulmonary circulation that is characterized by high pressure and low flow because of both passive and active elevation of PVR (Figure 48-5, A). The passive resistance most likely is related to compression of pulmonary capillaries by fetal lung liquid, but there is also a high degree of active vasomotor tone resulting from various mediators and hypoxic stimuli. Thus PVR exceeds systemic vascular resistance (SVR), resulting in right atrial and ventricular pressures exceeding left atrial and ventricular pressures. High PVR results in right-to-left shunting of blood across the foramen ovale, and most of the blood ejected by the right ventricle flows across the ductus arteriosus into the descending aorta. Persistence of elevated PVR after birth without the benefit of placental oxygenation results in the profound hypoxemia that characterizes PPHN.
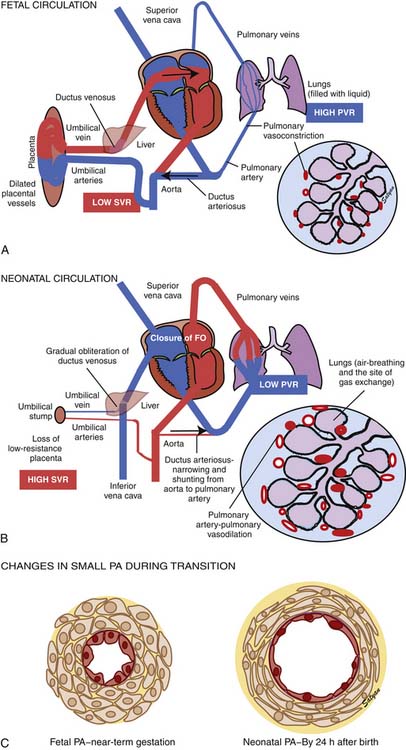
The second stage of normal transition is accomplished when the fluid-filled fetal lungs are distended with air during the first breath (Figure 48-5, B).20 A rapid decrease in PVR occurs with mechanical distension of the pulmonary vascular bed. The entry of air into the alveoli improves oxygenation of the pulmonary vascular bed, further decreasing PVR.45 At birth PVR decreases dramatically, which leads to an eightfold increase in Qp. The increase in Qp raises left atrial pressures above right atrial pressures, closing the foramen ovale. SVR increases at birth, in part because of removal of the low resistance bed of the placenta. As PVR becomes less than systemic, flow across the ductus reverses. Within the first 5 minutes after birth, oxygen-induced vasodilation and lung expansion decrease PVR to approximately half of systemic resistance. Over the first few hours after birth, the ductus arteriosus closes, largely in response to the increase in oxygen tension. At this point the normal postnatal circulatory pattern is established.
The third stage of the transitional circulation occurs for 12 to 24 hours after birth and accounts for the greatest reduction in PVR. In the final phase of neonatal pulmonary vascular transition, further decline in PVR is accompanied by rapid structural remodeling of the entire pulmonary bed from the main pulmonary arteries to the capillaries.46 During this remodeling, changes in the shape and geometric orientation of endothelial and smooth muscle cells cause luminal enlargement. Maturation of smooth muscle function, thinning of endothelial cells (Figure 48-5, C), and more gradual changes in elastic and connective tissue occur during the next few weeks.
Factors Responsible for Decrease in Pulmonary Vascular Resistance at Birth
The onset of ventilation with rhythmic inflation of the lungs at birth with a resultant increase in oxygen tension in the lungs leads to a decrease in PVR. Each of these stimuli has been shown to decrease vascular resistance and increase blood flow in the lungs of fetal lambs.45 Increasing oxygen tension alone by using a hyperbaric chamber decreased PVR and increased Qp by tenfold in mature fetal lambs.17 Inflation of lungs with gas may decrease resistance, at least in part by mechanical effects. Oxygen may dilate in part by direct effects on vascular endothelial and smooth muscles.20,47
The drop in PVR soon after birth is accompanied by production of prostacyclin (PGI2) and NO. Arachidonic acid metabolites such as PGs, generated through the cyclooxygenase pathway, are potent pulmonary vasodilators in the fetus. PGI2 synthesized by endothelial cells appears to relax smooth muscle by producing cyclic adenosine monophosphate (cAMP). PGI2 and its metabolites are more potent vasodilators than PGE2. Even though blockade of PG synthesis, either by indomethacin48,49 or meclofenamate,23 blunts the decrease in PVR, it does not completely disrupt the transition to gas exchange. In addition, cyclooxygenase through the PGE2 pathway plays an important role in maintaining patency of the ductus arteriosus. PPHN has been observed in infants of mothers receiving aspirin or nonsteroidal antiinflammatory drugs (NSAIDs) that inhibit cyclooxygenase activity.50 In this situation, the inhibitor acts through prenatal constriction of the ductus arteriosus or by decreasing PGI2 synthesis at birth.51 The cAMP signal transduction pathway is shown in Figure 48-6.
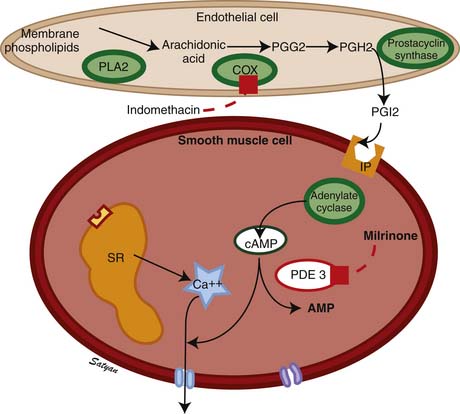
Acetylcholine,52 bradykinin,53 and histamine54 are fetal pulmonary vasodilators, which in many species act by an endothelium-dependent mechanism (see Figure 48-4). They stimulate the production of NO by vascular endothelium. NO activates soluble guanylate cyclase to produce the second messenger cyclic guanosine monophosphate (cGMP). cGMP induces relaxation of vascular smooth muscle through activation of a cGMP-dependent protein kinase that produces a lowering of cytosolic ionic calcium, in part through activation of potassium channels.55 There is strong evidence that NO is an important mediator of the decrease in PVR at birth. NO is a potent dilator of the fetal pulmonary circulation.56 The dilation of the fetal pulmonary circulation caused by an increase in oxygen tension is mediated in large part by endogenous synthesis of NO.57 In late gestation lambs, prolonged administration of nitric oxide synthase (NOS) inhibitors, which blocks endogenous NO synthesis, does not affect basal PVR but markedly blunts the decrease in PVR observed at birth.58
Evidence suggests that many other mediators can act as pulmonary vasodilators during fetal life and at birth. The purines adenosine triphosphate and adenosine are potent pulmonary vasodilators in the fetal lamb that may also be involved at birth.59–63 Natriuretic peptides such as atrial natriuretic peptide, B-type natriuretic peptide, and C-type natriuretic peptide dilate fetal pulmonary vasculature by increase cGMP through particulate guanylate cyclase.64 Arachidonic acid metabolites such as epoxyeicosatrienoic acids, which are generated through the cytochrome P450 pathway, are potent pulmonary vasodilators.65 Because pulmonary vasodilation at birth is a vital step in establishing postnatal life, it is logical that there would be sufficient redundant vasodilators to compensate for failure or inadequacy of any single pathway.37
Persistent Pulmonary Hypertension of the Newborn
PPHN is a serious clinical condition that can result from diverse etiologies and is characterized by failure of the pulmonary circulation to adapt to extrauterine life.
Pathophysiology
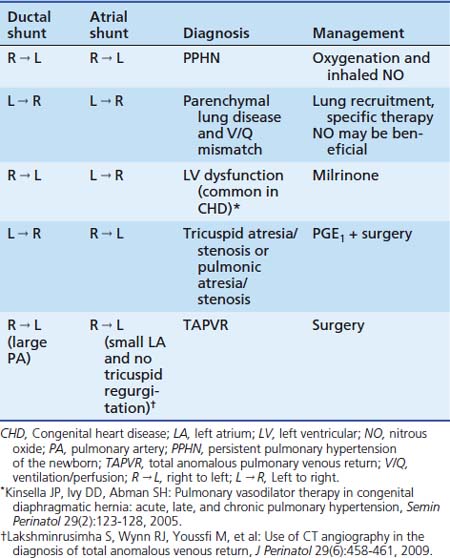
Elevated pulmonary to systemic vascular resistance ratio (PVR/SVR) resulting from either vasoconstriction, structural remodeling of the pulmonary vasculature, intravascular obstruction, or lung hypoplasia (Figure 48-7) characterizes PPHN. This leads to right-to-left shunting of blood across the foramen ovale and ductus arteriosus, resulting in hypoxemia. Numerous disease states with diverse etiologies can result in a similar final pathophysiology. About 10% of cases with PPHN are idiopathic, with no associated pulmonary airspace pathology. However, PPHN is usually associated with other acute respiratory conditions, such as MAS, respiratory distress syndrome (RDS), pneumonia, or congenital diaphragmatic hernia (CDH) (Figure 48-8). Hypoxemia in these conditions can be due to ventilation/perfusion (V/Q) mismatch and intrapulmonary, as well as extrapulmonary, right-to-left shunting of blood. In some newborns with hypoxic respiratory failure, a single mechanism predominates (e.g., extrapulmonary right-to-left shunting in idiopathic PPHN). However, more commonly, several of these mechanisms contribute to hypoxemia. In MAS, obstruction of the airways by meconium results in decreasing V/Q ratios and increasing intrapulmonary right-to-left shunt. Other segments of the lungs may be overventilated relative to perfusion, causing increased physiologic dead space. The same patient also may have severe PPHN with extrapulmonary right-to-left shunting at the ductus arteriosus and foramen ovale. The PPHN in patients with MAS may result from the alveolar hypoxia, from inflammatory mediators, or from abnormal pulmonary vascular muscularization.
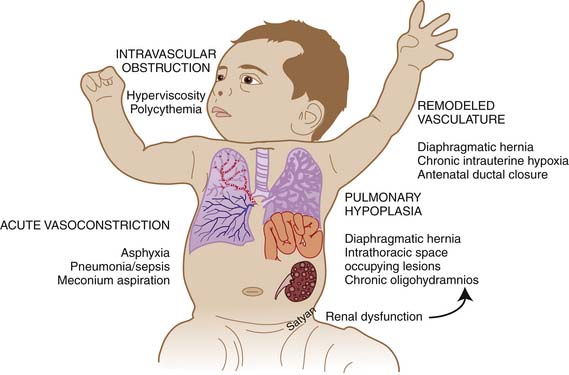
Figure 48–7 Mechanisms of persistent pulmonary hypertension of the newborn.
(Copyright Satyan Lakshminrusimha.)
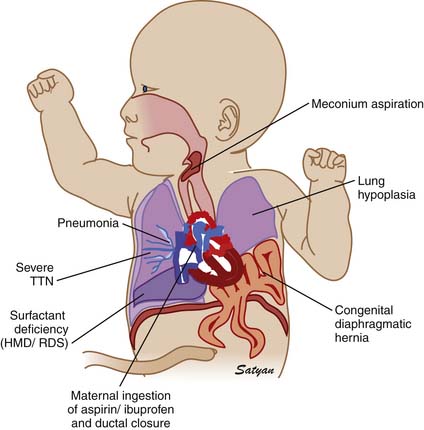
Figure 48–8 Clinically common conditions associated with persistent pulmonary hypertension of the newborn.
(Copyright Satyan Lakshminrusimha.)
Pneumonia or meconium aspiration may release inflammatory mediators that induce vasoconstriction. Vasoconstrictors such as leukotrienes, platelet-activating factor, thromboxanes,66 and ET-167 have been found to be elevated in patients with PPHN. Chronic intrauterine ETA receptor blockade following ductal ligation decreases pulmonary arterial pressure in utero and decreases RV hypertrophy, and distal muscularization of small pulmonary arteries increases the fall in PVR at delivery in newborn lambs with PPHN.39 Thus ET-1 acting through the ETA receptor stimulation might contribute to the pathogenesis and pathophysiology of PPHN. Derangements in the NO pathway of vasodilation also can result in the physiologic characteristics of PPHN. Pulmonary endothelial nitric oxide synthase (eNOS) gene and protein expression and enzyme activity are decreased in fetal lambs with PPHN induced by antenatal ductal ligation.68 In addition, the response to stimulators of eNOS is lost.69 In these lambs with PPHN, the vascular response to NO itself is also diminished,70 whereas the response to cGMP is normal. Thus the decreased responsiveness appears to result from decreased vascular smooth muscle sensitivity to NO at the level of soluble guanylate cyclase. The cGMP pathway of signal transduction is shown in Figure 48-4. Because NO both vasodilates and inhibits vascular smooth muscle growth, diminished eNOS expression may contribute to both abnormal vasoreactivity and excessive muscularization of pulmonary vessels in patients with PPHN.
PH sometimes occurs because of an abnormal pulmonary vascular bed despite the absence of alveolar hypoxia and hypercapnia and of lung inflammation. These infants can be grouped according to the degree of muscularization and the number of pulmonary arteries.71 In infants with hypoplastic lungs, as in those with congenital heart disease (CHD) and oligohydramnios sequence, PPHN may arise primarily as a consequence of a decreased number of vessels causing a decreased cross-sectional area of the pulmonary vascular bed, leading to flow restriction (see Figure 48-7). Patients with alveolar capillary dysplasia may have a similar vascular hypoplasia. These cases may be complicated by increased muscularization of the vessels.
Antenatal exposure to NSAIDs such as aspirin and ibuprofen has been associated with PPHN.50 More recently, an association between the use of selective serotonin reuptake inhibitor class of antidepressants after 20 weeks of gestation and PPHN has been found.72,73 Prenatal exposure to fluoxetine (a selective serotonin reuptake inhibitor) induced fetal PH in rats.74 The exact mechanism of this association is not clear. Polycythemia and hyperviscosity also may increase PVR and contribute to PPHN. Similarly, hypothermia, acidosis, and hypoxemia can aggravate PPHN.
Clinical Presentation
Labile hypoxemia is the hallmark of PPHN (Figure 48-9). A newborn infant who is extremely labile, with frequent desaturation episodes and wide swings in arterial PO2 without changes in ventilator settings, should suggest the possibility of PPHN. Lability also can occur in the face of significant parenchymal disease when V/Q mismatch is severe. Auscultation of the heart in babies with PPHN may reveal a single S2, which can be loud, and a systolic murmur of tricuspid regurgitation. Chest radiograph findings may vary depending on the etiology of PPHN. Hypoxemia out of proportion to the degree of parenchymal disease severity on chest radiography should suggest PPHN. Measurement of preductal and postductal arterial oxygenation can confirm PPHN. A difference in arterial PO2 20 mm Hg or greater or oxygen saturation 10% or greater should be considered significant. Minimal or no difference in oxygen tension does not exclude PPHN because shunting at the atrial level produces no ductal gradient and probably is the most common site of shunting. Thus in clinical practice, the gold standard in defining PPHN rests on the echocardiographic findings of right-to-left shunting of blood at the foramen ovale and/or the ductus arteriosus, as well as estimates of PAP. Doppler measurements of atrial and ductal level shunts provide essential information when managing a newborn with hypoxic respiratory failure.75 For example, left-to-right shunting at the foramen ovale and ductus arteriosus with marked hypoxemia suggests predominant intrapulmonary shunting, and interventions should be directed at optimizing lung inflation. Similarly, presence of right-to-left shunting at the ductal level and left-to-right shunting at the atrial level suggests PPHN with left ventricular dysfunction with some pulmonary venous hypertension (see Table 48-1). This finding may be associated with CHD.76
Treatment
General Measures
Understanding the dynamic pathophysiology underlying right-to-left shunting is important to the successful management of PPHN. In patients with PPHN, even mild stress can cause PO2 to plummet within minutes. Vigorous and persistent resuscitative measures are often necessary to promote pulmonary vasorelaxation. Accordingly, neonates and children with PPHN are often very sensitive to activity and agitation. Minimal stimulation and sedation with narcotics such as fentanyl and morphine are commonly used to achieve this goal. However, paralysis should be avoided because it is associated with increased mortality.77 The lability in PaO2 may result from the fact that when pulmonary and systemic arterial pressures are similar, small alterations in the ratio of the two can produce large changes in extrapulmonary shunting (see Figure 48-8).78 Systemic blood pressures should be maintained at a high normal range for age and gestation, because an increased systemic resistance may decrease the degree of right-to-left shunting. Hypotension resulting from hypovolemia should be treated aggressively with volume replacement. If hypotension persists despite volume replacement, inotropic support with dopamine, dobutamine, and epinephrine may be required. All patients with PPHN benefit from a core group of therapies that includes management of hypothermia, hypocalcemia, acidosis, hypoglycemia, and polycythemia. In cases of PPHN resulting from perinatal asphyxia, correcting alveolar hypoxia, hypercarbia, and metabolic acidosis with administration of oxygen, conventional ventilation, and a buffer should restore normal pulmonary vasodilation. Surprisingly, few prospective randomized trials have been conducted of most of the therapeutic modalities advocated for the treatment of PPHN. Older therapies such as hyperventilation and alkalosis were introduced into clinical practice based on animal studies or short-term studies that included very small numbers of patients and used physiologic response rather than patient outcome as the end point. Only the newer therapies of inhaled nitric oxide (iNO) and extracorporeal membrane oxygenation (ECMO) have been rigorously evaluated in controlled clinical trials. In a National Institute of Child Health and Human Development (NICHD) observational study,77 the use of hyperventilation varied among the 12 NICHD centers from 33% to 92%, and alkali infusion ranged from 27% to 93%. Similar variation was seen in the use of inotropic agents (46% to 100%) and intravenous vasodilators (13% to 81%). High-frequency ventilation (HFV) use ranged from 0% to 73%, and ECMO use varied from 0% to 85%. Some of the variation is expected given the wide range of pathophysiology contributing to PPHN. A more likely explanation for the wide variation seen, however, is the overall lack of proven efficacy of many of these therapies.
Hyperventilation and Alkali Infusion
Animal studies documented the sensitivity of the pulmonary vasculature to both hypoxia and acidosis.79 Short-term studies during cardiac catheterization showed reductions in PAP and elevation of oxygen tension following hyperventilation.78,80 Based on these studies, hyperventilation became the mainstay of ventilation of term infants with hypoxic respiratory failure and PPHN. In some patients the benefits of hyperventilation may be outweighed by risks of barotrauma or volutrauma. Moreover, subsequent observations have raised concern of impaired cerebral perfusion and neurosensory deafness at extremes of alkalosis.81,82 Studies of infants with PPHN maintaining normal PCO2 (40 to 60 mm Hg) indicate similar or better outcomes and with less chronic lung disease.83,84 Many neonatologists have moved away from the practice of hyperventilation in neonates with PPHN. Animal studies have shown that the beneficial effects of hyperventilation result from altered pH rather than from changes in PCO2 or minute ventilation.85,86 At one time alkali infusion to maintain alkaline pH to induce pulmonary vasodilation was a standard practice. In the aforementioned NICHD observational study, the group treated with alkali had a greater chance of treatment with ECMO compared with those treated with hyperventilation and an increased rate of supplemental oxygen at 28 days.77 Alkali infusion with bicarbonate increases CO2 production, necessitating higher ventilator support. Lack of patient outcome data with both hyperventilation and alkali infusion and availability of better therapeutic options have led to less use of these outdated management strategies.
Oxygen
Oxygen is a specific and potent pulmonary vasodilator, and increased oxygen tension is an important mediator of reduction in PVR at birth. Alveolar hypoxia and hypoxemia increase PVR and contribute to the pathophysiology of PPHN. Avoiding hypoxemia by mechanical ventilation with high concentrations of oxygen continues to be the mainstay of PPHN management. However, exposure to hyperoxia may result in formation of oxygen free radicals and lead to lung injury. Recent evidence suggests that brief exposure to 100% oxygen in newborn lambs results in increased contractility of pulmonary arteries87 and reduces response to inhaled NO.88,89 The biological half-life of endogenous NO is related to the local concentration of superoxide anions (see Figure 48-4).90 Administration of intratracheal recombinant human superoxide dismutase (an antioxidant that breaks down superoxide anions) results in improved oxygenation in lambs with PPHN.91,92 Based on these studies, it appears that avoiding hyperoxia is as important as avoiding hypoxia in the management of PPHN.
The optimal PaO2 in the management of PPHN is not clear. Wung and colleagues84 have suggested that gentle ventilation with avoidance of hyperoxia and hyperventilation results in good outcome in neonates with respiratory failure. Decreasing PaO2 below 45 to 50 mm Hg results in increased PVR in newborn calves79 and lambs.89 In contrast, maintaining PaO2 greater than 70 to 80 mm Hg does not result in additional decrease in PVR in both control lambs and lambs with PPHN. In animal studies, hypoxemia results in pulmonary vasoconstriction and normoxemia reduces PVR but hyperoxemia does not result in additional pulmonary vasodilation. However, to date, randomized studies comparing different PaO2 targets have not been conducted in infants with PPHN.
Lung Recruitment Strategies
Surfactant
Clinical reports suggest that surfactant therapy improves oxygenation in term infants with RDS, pneumonia, and MAS.93,94 Surfactant administration results in reduced need for ECMO in infants of 36 weeks or greater gestation at birth. Near-term and term infants delivered by elective repeat cesarean section may be one subset of patients who are at risk for deficiency or dysfunction of surfactant and progressive hypoxic respiratory failure.95 If the cause of PPHN is parenchymal lung disease such as RDS, meconium aspiration or pneumonia, exogenous surfactant may be beneficial in improving oxygenation and reducing the need for ECMO.94
High-Frequency Ventilation
Many clinicians use HFV to manage infants with PPHN. Considering the important role of parenchymal lung disease in specific disorders resulting in PPHN, adequate lung inflation and optimal ventilation are as essential as pharmacologic vasodilator therapy. In the case of inhaled vasodilators, optimal inflation and ventilation may be necessary for drug delivery.96 Infants with PPHN from a variety of causes have been successfully treated with HFV.97 High-frequency oscillatory ventilation (HFOV) decreases PaCO2 and increases oxygenation in infants with PPHN. HFOV may improve oxygenation through safer use of higher mean airway pressures to maintain lung volume and prevent atelectasis. Two studies have evaluated the effectiveness of HFV compared with conventional ventilation in rescuing infants with respiratory failure and PPHN from potential ECMO therapy.98,99 Neither mode of ventilation was more effective in preventing ECMO in these infants. In clinical pilot studies using iNO, combination of HFOV and iNO resulted in the greatest improvement in oxygenation in some newborns who had severe PPHN complicated by diffuse parenchymal lung disease and underinflation.100 A randomized controlled trial demonstrated that treatment with HFOV and iNO was often successful in patients who failed to respond to HFOV or iNO alone in severe PPHN, and the differences in responses were related to the specific disease associated with PPHN. Infants with RDS and MAS benefit most from a combination of HFOV and iNO therapy.101,102
Nitric Oxide
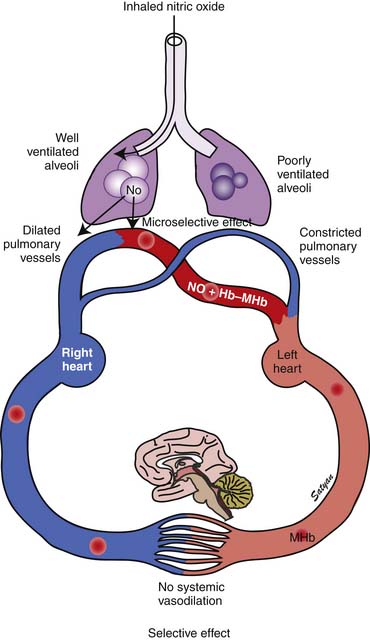
In 1996 the Food and Drug Administration (FDA) approved iNO for use in neonates who are 35 weeks’ gestation and older with PPHN. The physiologic rationale for using iNO for treatment of PPHN103,104 is based on its ability to achieve potent and selective pulmonary vasodilation without decreasing systemic vascular tone (Figure 48-9). Once iNO enters the intravascular space, it combines with hemoglobin to form methemoglobin and does not exert a vasodilator effect on the systemic circulation. Inhaled NO also exerts a microselective effect and reduces V/Q mismatch. Being an inhaled vasodilator, NO enters only ventilated alveoli and redirects pulmonary blood by dilating adjacent pulmonary arterioles and reduces V/Q mismatch (Figure 48-10). Studies in newborn lambs have shown that prolonged administration of NO increased survival rates without increasing the incidence of acute lung injury in lambs with PPHN.105
Three randomized trials on the use of iNO in newborns with PPHN and respiratory failure were published in 1997. In a randomized controlled clinical trial of patients with PPHN reported by Roberts and colleagues,106 oxygenation doubled in 58% of treated patients in response to 80 ppm iNO. In addition, twice the proportion of the treated group avoided ECMO compared with the control group. In the Neonatal Inhaled Nitric Oxide Study, 235 infants older than 34 weeks’ gestation who were diagnosed with hypoxic respiratory failure were randomly assigned to receive 20 ppm iNO or were assigned to a control group. Infants whose partial pressure of arterial oxygen increased by 20 mm Hg or less were studied for a response to 80 ppm iNO or control gas. The end point of this trial was death or ECMO. Although the mortality rate was no different in either treatment arm, there was a 40% reduction in the need for ECMO among infants treated with iNO compared with control subjects.107 Only 6% of infants who failed to respond to treatment with 20 ppm of iNO responded to the higher dose in this study. In a study by Kinsella and colleagues,102 205 infants with PPHN were randomly assigned to receive iNO and conventional ventilation or to receive HFOV alone. Those who did not respond to either therapy received the combination of iNO and HFOV. Treatment with HFOV in combination with iNO was successful in some patients who did not respond to one treatment alone. The differences were partly related to the specific disease (RDS and MAS) associated with PPHN.102 Because adequate lung inflation appears to be necessary for optimal response to iNO, lung recruitment strategies with HFOV should augment the response to iNO. Clark and colleagues108 reported a 38% reduction in ECMO and no difference in mortality among 248 infants randomly assigned to receive 20 ppm iNO for 24 hours followed by 5 ppm for no more than 96 hours. A meta-analysis of the results of seven randomized trials of iNO use in newborns with PPHN demonstrated that 58% of hypoxic near-term and term infants responded to iNO within 30 to 60 minutes.109 Mortality was not reduced in any of the NO studies analyzed, but use of ECMO as a rescue therapy in nonresponders was significantly decreased.
Inhaled NO by itself is toxic at higher concentrations. Potential adverse effects include methemoglobinemia, pulmonary edema, and platelet dysfunction. NO reacts with superoxide anion to form peroxynitrite, which causes lipid peroxidation and other oxidative injury to cell membranes. NO2 is even more toxic. Careful monitoring of both NO and NO2 levels during administration is mandatory. Because the optimal dosing and timing of iNO administration remains unclear and the potential toxicities are dose-related, lower doses might afford both safety and efficacy in the management of these infants. Two studies using 2 ppm iNO yielded contradictory results. In one study, 2 ppm iNO diminished the clinical response to 20 ppm.110 In the other study, the initial exposure to a very low dose did not compromise the response to higher doses.111 In a study involving direct measurements of PAP during cardiac catheterization, iNO produced peak improvement in oxygenation at 5 ppm, whereas peak improvement in the pulmonary-to-systemic arterial pressure ratio did not occur until an iNO dose of 20 ppm, which suggests that an initial dose of 20 ppm is optimum for the treatment of PPHN.112 The use of higher doses of iNO is associated with increased methemoglobin levels, especially at 80 ppm but not at 40 or 20 ppm.113 The results of randomized controlled trials support the use of iNO at starting does of 20 ppm in near-term and term infants. In summary, iNO offers substantial benefit to a large proportion of near-term and term newborns with hypoxic respiratory failure who do not respond to ventilatory support, lung recruitment, and oxygen.
The timing of initiation of iNO in patients with hypoxic respiratory failure is not clear. Severity of hypoxemia in neonates is measured by oxygenation index (OI = Mean airway pressure in cm of water × FIO2 × 100/PaO2 [in mm Hg]). An OI of 40 is often used as an indication for ECMO therapy. An OI of 25 is associated with a 50% risk of requiring ECMO or dying.37 Thus the acceptable indication for treatment with iNO include an OI greater than 25 with echocardiographic evidence of PPHN or a higher OI with or without evidence of right-to-left shunt. However, it has been suggested that initiating iNO at a lower OI may be associated with reduced need for ECMO.114 Konduri and colleagues115 randomly assigned neonates who were born at 34 weeks’ gestation or later, required assisted ventilation, and had an OI of 15 or greater and less than 25 to receive early iNO or to receive simulated initiation of iNO (control). Infants who had an increase in OI to 25 or more were given iNO as standard therapy. Arterial oxygen tension increased by more than 20 mm Hg in 73% of infants who received early iNO (n = 150) and in 37% of infants in the control group (n = 149) after study gas initiation. Infants in the control group received standard iNO and deteriorated to an OI score greater tha 40 more often than did infants who were given early iNO. The incidence of death (early iNO group, 6.7% vs. control group, 9.4%), ECMO (10.7% vs. 12.1%), and their combined incidence (16.7% vs. 19.5%) were similar in both groups.115 Follow-up evaluations showed no differences between the 2 groups in the incidence of neurodevelopmental impairment (early iNO group, 27%; control group, 25%) and hearing impairment (early iNO group, 23%; control group, 24%). Mental development index scores were similar in the two groups; however, psychomotor developmental index scores were significantly higher in the control group (early iNO group, 89 ± 17.7; control group, 93.5 ± 18.4).116 These findings suggest that caution must be exercised while initiating iNO at lower OI levels.
Inhaled NO should be weaned gradually to avoid rebound PH.117 A flow diagram showing the protocol for weaning iNO at The Women and Children’s Hospital of Buffalo is shown in Figure 48-11.
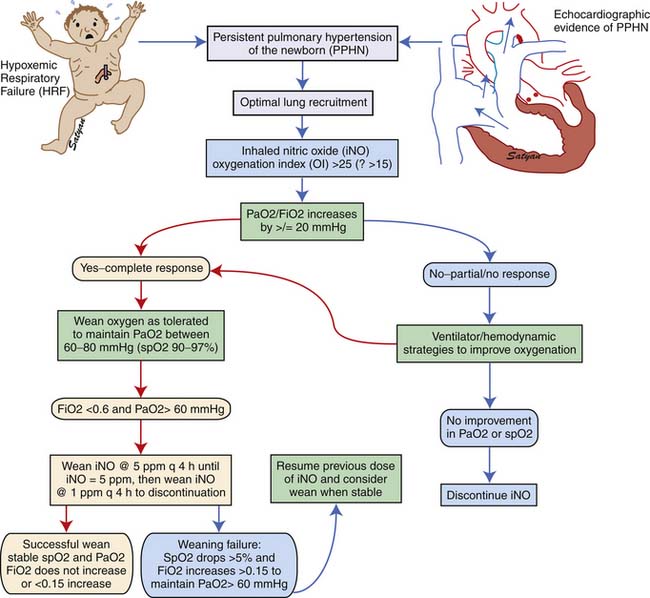
Figure 48–11 Weaning protocol for inhaled nitric oxide in use at The Women and Children’s Hospital of Buffalo, NY.
The outcome of infants treated with iNO collectively supports both the efficacy and safety of this mode of treatment. Reports identify significant medical and neurodevelopmental sequelae of PPHN with or without iNO and point out the necessity for coordinated multidisciplinary follow-up for these infants. The overall rate of neurodevelopmental handicap in infants treated with NO was 46%, with 25% mildly affected and 21% severely affected at age 1 year in one study.118 In another study, mild and severe neurodevelopmental handicaps were 14% and 12% at age 1 year and 9% and 12% at age 2 years.119 In these studies sensorineural hearing loss was present in 6% to 19% of infants with PPHN treated with iNO. No difference was noted between control and treatment groups for these outcomes. Published reports on the use of iNO in ECMO centers have not substantiated early concerns that iNO would adversely affect outcome by delaying ECMO utilization. Inhaled NO treatment may play an important role in stabilizing patients before ECMO is initiated, thus improving the chances of ECMO cannulation without further clinical deterioration. The Committee on the Fetus and Newborn of the American Academy of Pediatrics has suggested that iNO use be limited to tertiary care centers where ECMO is available.120 In non-ECMO centers, a system should be in place to continue iNO during transport even if a response occurs, because not all physiologic responders avoid ECMO. For the same reason, the combination of HFOV and iNO should be used cautiously in non-ECMO centers.
Phosphodiesterase Inhibitors
Vasodilators such as NO and prostacyclin relax vascular smooth muscle by increasing intracellular concentrations of the second messengers cGMP (see Figure 48-4) and cAMP, respectively (see Figure 48-5). However, nearly 40% of patients with PPHN do not demonstrate a sustained improvement in oxygenation with this management. Because iNO is not universally effective, there has been considerable interest in understanding and targeting other biochemical pathways that regulate pulmonary vasoconstriction in PPHN. Inhibition of the cGMP degrading phosphodiesterase (PDE5) by sildenafil and inhibition of the cAMP degrading phosphodiesterase (PDE3) by milrinone offer additional tools to achieve pulmonary vasodilation in patients with PPHN. Two studies have evaluated the use of oral sildenafil in PPHN.121,122 In centers lacking iNO therapy, oral sildenafil (dose range 1 to 3 mg/kg every 6 hours) has been shown to improve oxygenation and reduce mortality. A recent study showed that intravenous (IV) sildenafil was effective in improving oxygenation in patients with PPHN with and without prior exposure to iNO.123 Systemic hypotension was the most common adverse effect. Administration of a loading dose slowly over 3 hours followed by a maintenance dose of sildenafil reduced the risk of systemic hypotension. These data suggest a beneficial effect for oral as well as IV sildenafil in patients with PPHN. Because oral or IV agents are not selective to pulmonary circulation, caution must be exercised about the risk of systemic hypotension with oral sildenafil. IV sildenafil is not available currently in the United States, but the same caution regarding systemic hypotension exists. The use of oral sildenafil for treatment of PH is approved by the FDA for adults but not for children.
Milrinone is the prototype PDE3 inhibitor and is commonly used in adult and pediatric intensive care settings as an inotropic vasodilator. Agents that increase cAMP levels in pulmonary arterial smooth muscle cells (such as milrinone and prostacyclin) provide an alternate pathway of pulmonary vascular relaxation and potentially result in improved oxygenation in patients with poor response to iNO. In newborn lambs with PPHN, milrinone effectively relaxes pulmonary arteries124 and improves oxygenation.125 There is a marked increase in PDE3 activity and reduced cAMP in pulmonary arteries following ventilation of lambs with iNO.126 Milrinone can be effective in PPHN by inhibiting PDE3 and increasing cAMP, causing direct pulmonary vasodilation and a synergistic effect with iNO. Milrinone may also improve cardiac function by positive inotropy (improved contraction), lusitropy (improved relaxation), and reduced ventricular afterload. Bassler and colleagues127 and McNamara and associates128 described 13 patients with PPHN refractory to iNO from Ontario, Canada, who were treated effectively with intravenous milrinone. This therapy is associated with a risk of hypotension and intraventricular hemorrhage and is not currently approved by the FDA.
Extracorporeal Membrane Oxygenation
If the heart and lungs cannot support the newborn, they can be bypassed with ECMO. Several randomized trials have indicated improved survival of infants supported with ECMO. In a large prospective trial conducted in the United Kingdom, 121 infants with severe respiratory failure were randomly assigned to ECMO or conventional management.129 Survival in the patients treated with ECMO was significantly greater than in the control group (68% vs. 41%). Neurologic outcome was similar among survivors of either treatment arm, indicating that ECMO likely did not contribute to morbidity in this group of critically ill infants. ECMO has been shown to be both clinically129 and economically130 justifiable for mature newborn infants with severe respiratory failure. ECMO is not a specific treatment for any disease but rather a method of supportive treatment, in which the patient is kept alive while the lungs and their vasculature recover. As newer treatment modalities including HFOV, surfactant therapy, and NO have become available for treatment of hypoxemic respiratory failure, ECMO use in newborns has decreased considerably.131,132 Because of serious inherent risks, such as systemic and intracranial hemorrhage, for infants with PPHN, the procedure presently is reserved for newborn infants with reversible pulmonary disease in whom alternative therapies have failed. However, ECMO should be initiated before the infant is moribund.





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


